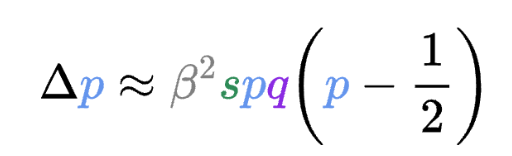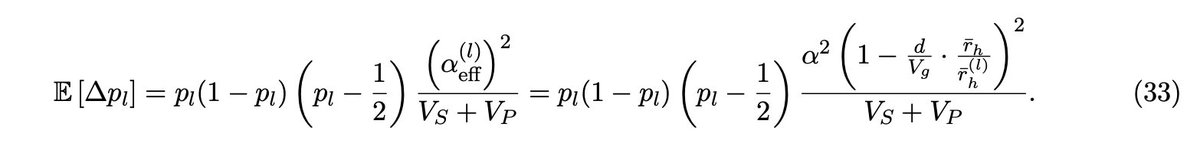Er, has anyone else realized that #claude is very happy to read your secrets and echo them back to you in plain text? e.g. it happily tells me all about a value in my .env file. Claude's exclusion rules don't actually work, see here for why and a solution
chriscole.substack.com/p/careful-clau…

English