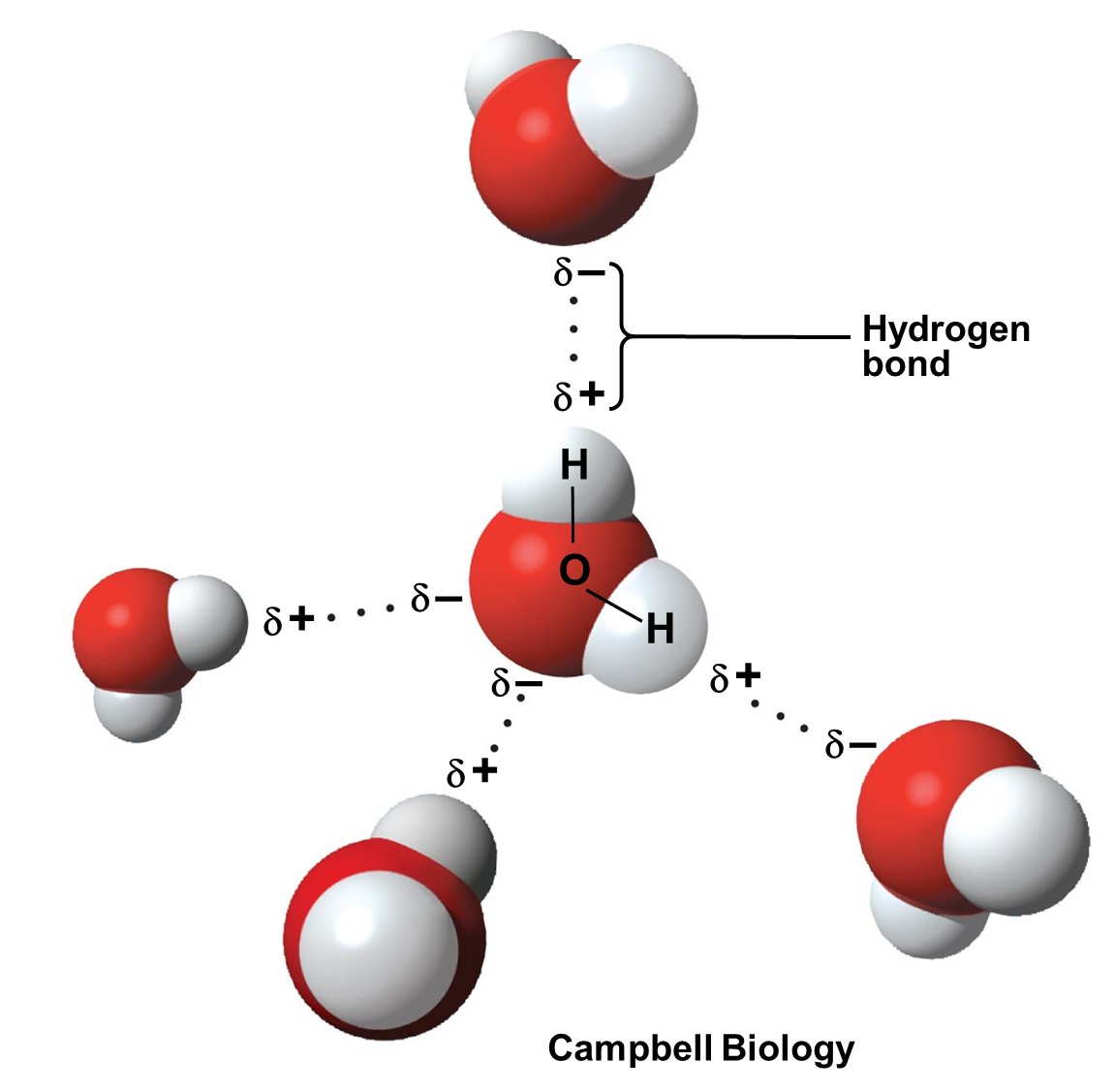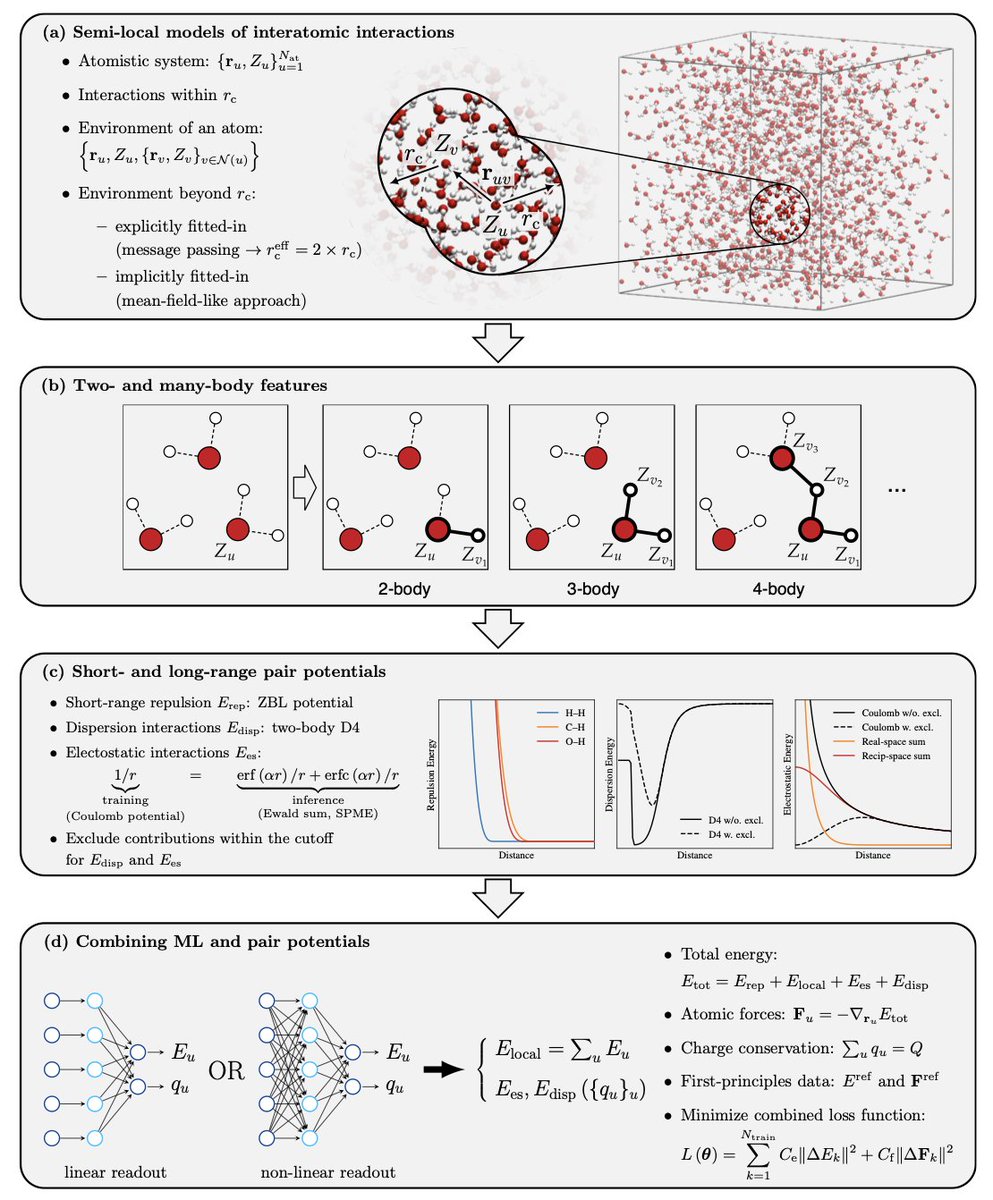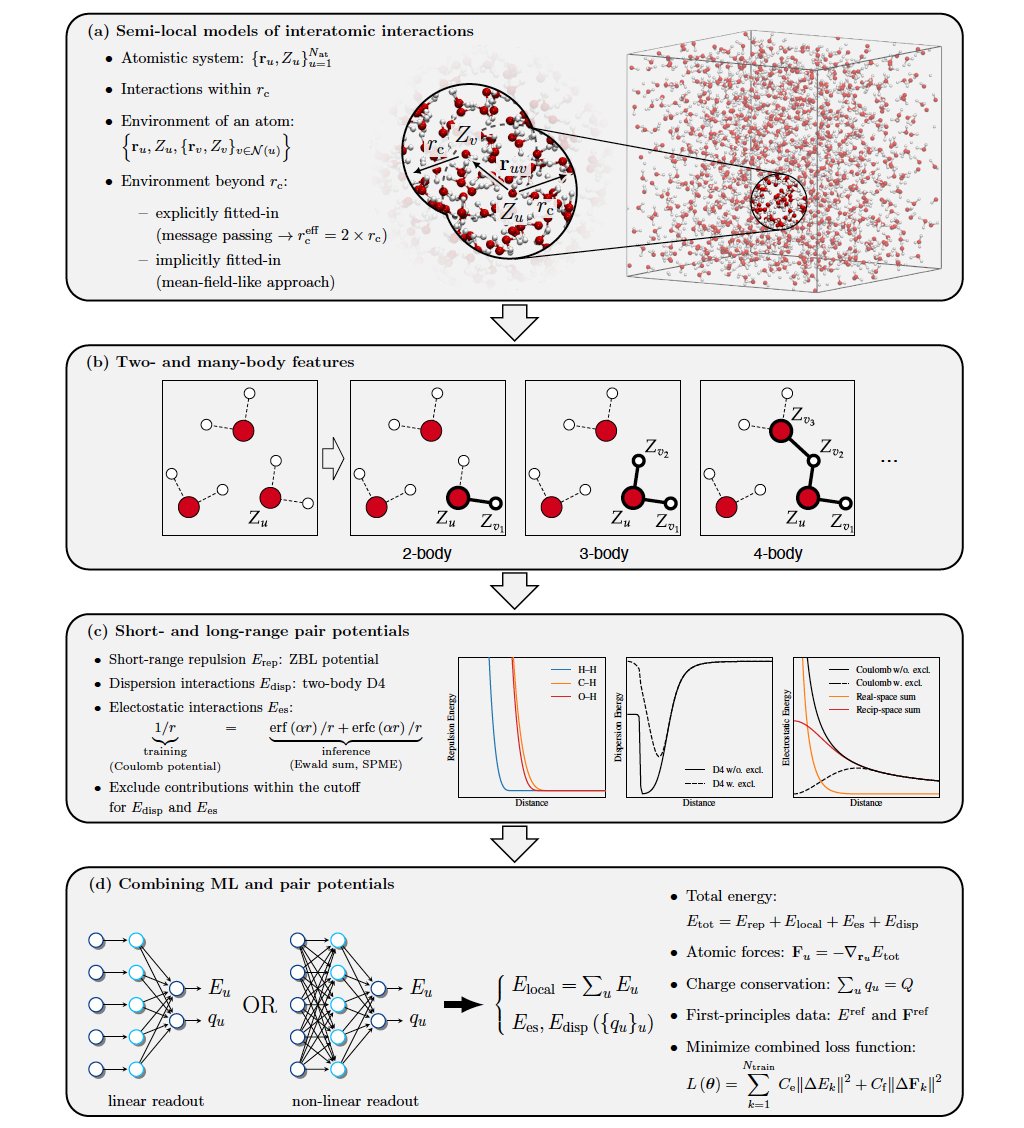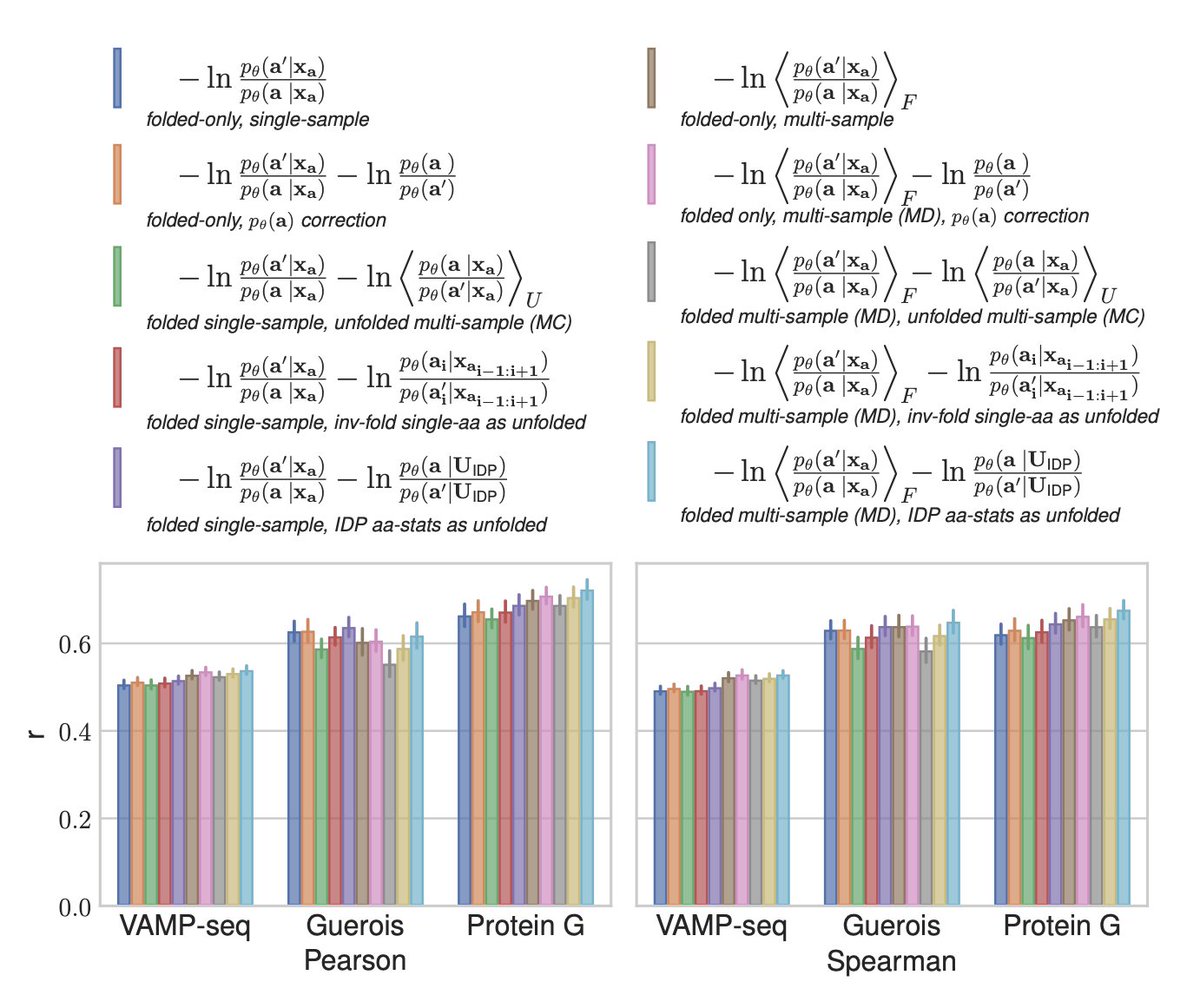
Angehefteter Tweet

Had a lot of fun lecturing on AI-based protein structure prediction at the STRUCTURAL BIOLOGY 2.0 workshop at the institute Pasteur Montevideo. It was great connecting with so many talented experimental and comp structural biologists @red_cebem @IUCr @ICGEB @IPMontevideo 🇺🇾

English