Manoj K Valluru retweetledi

Compressing the collective knowledge of ESM into a single protein language model @naturemethods
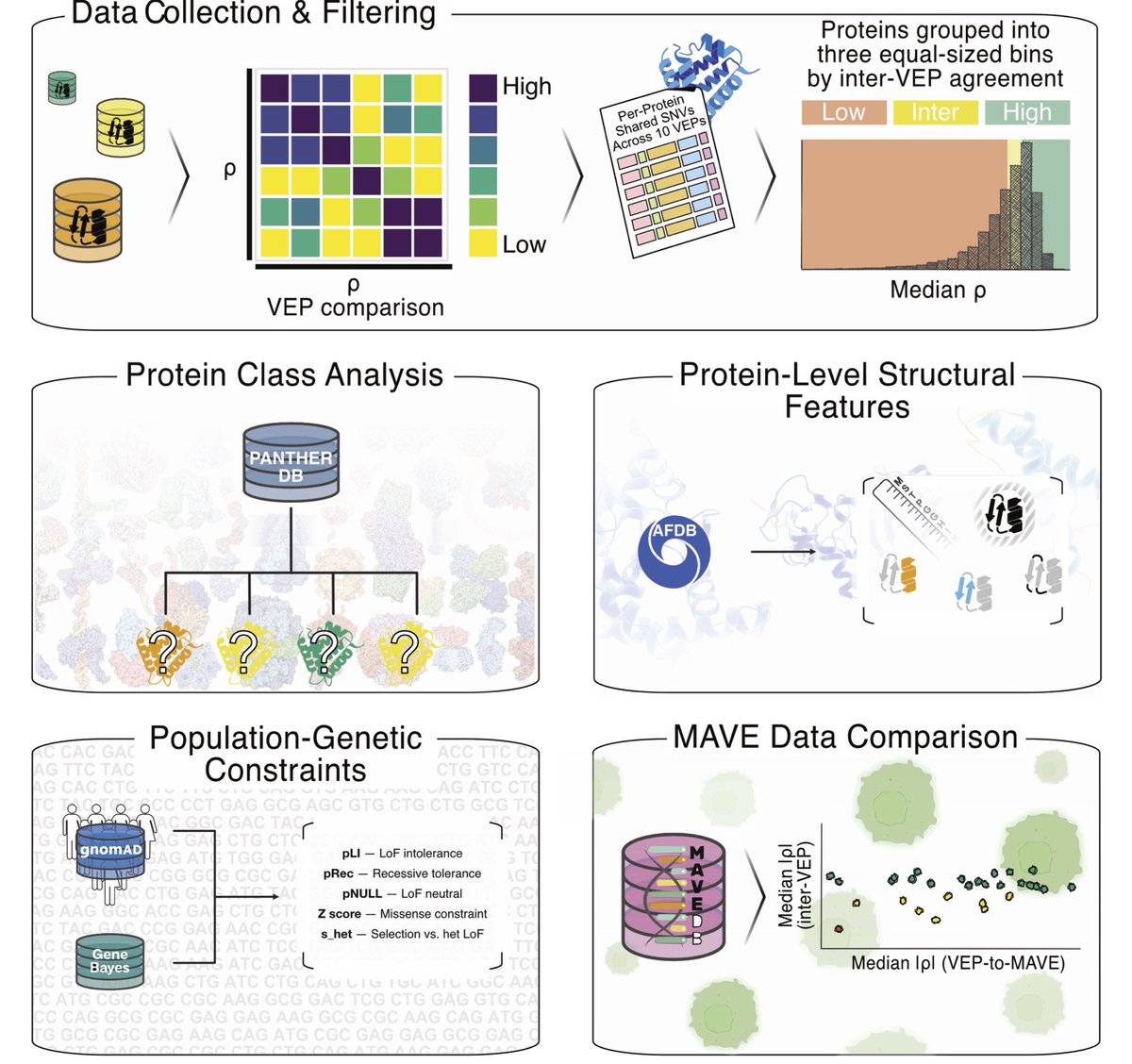
1. The paper argues that “sequence-only” protein language models (PLMs) are not intrinsically capped for variant-effect prediction (VEP); instead, their evolutionary signals are fragmented across model families and can be recovered by making models learn from each other.
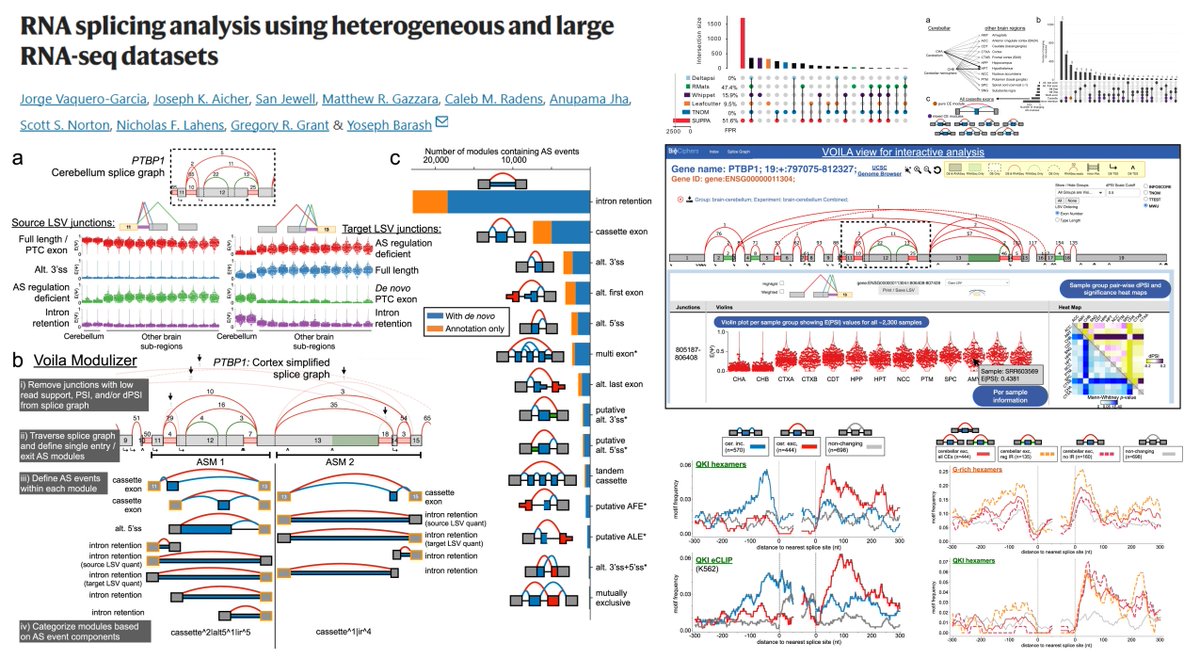
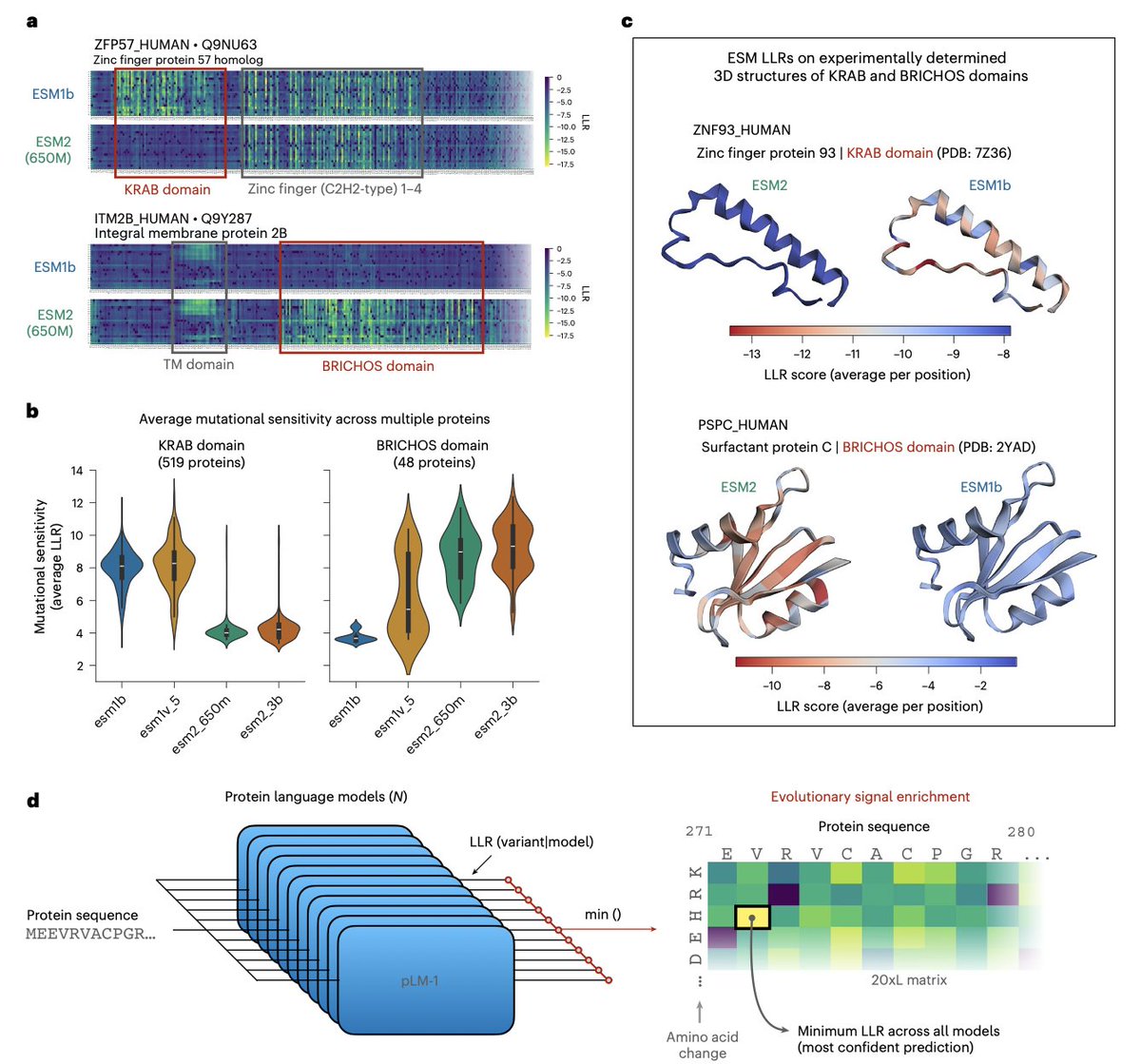
2. Key observation: closely related ESM models have complementary blind spots. For example, ESM2 models systematically miss KRAB-domain conservation signals, while ESM1b/ESM1v can miss BRICHOS-domain signals; yet at least one model in the family captures each domain’s mutational sensitivity.
3. They introduce a simple but effective ensemble rule: for each missense mutation, take the minimum log-likelihood ratio (LLR) across models (ESMIN), i.e., “maximum confidence” scoring. This can amplify subtle evolutionary constraints that averaging would dilute.
4. A theoretical analysis explains when min-LLR beats averaging: if pathogenic-variant LLRs are more dispersed across models than benign-variant LLRs (variance asymmetry). The ESM family empirically shows this property, making maximum-confidence aggregation advantageous.
5. ESMIN is evaluated using 11 sequence-only ESM models (ESM1b, five ESM1v, five ESM2; excluding ESM2-15B). It outperforms averaging-based ensembles and improves ProteinGym DMS correlations, with gains occurring in ~50% of assays (versus ~20% for typical ensembles).
6. Main methodological contribution: “maximum-confidence co-distillation.” For each protein, all models score all mutations; the element-wise minimum LLR matrix becomes a teacher signal, and each model is trained (variant-level MSE) to match these confident targets—without MSAs, structures, or population genetics features.
7. Co-distillation substantially improves every participating model, including small ones: ESM2-8M improves on ClinVar AUC from ~0.65 to ~0.88. Several co-distilled single models (e.g., ESM2-3B, ESM1b, ESM2-650M) can even surpass the ESMIN teacher signal (“student surpasses teacher”).
8. Robustness/ablation: improvements persist when training data are heavily reduced and de-homologized. With only ~1% of human proteins (~200 sequences; <30% identity to benchmark proteins), ESM2-35M reaches ~97% (ClinVar) and ~94% (DMS) of its peak co-distilled performance.
9. Iterative procedure: after round 1 (min-LLR co-distillation), additional rounds switch to average-aggregation co-distillation. As models improve, class-conditional variances become more symmetric, making averaging slightly better; after 3 rounds, a single 3B model matches the ensemble—named VESM-3B.
10. Practical compression: VESM-3B is distilled into smaller models (650M, 150M, 35M) that retain most performance (reported as >98% on Balanced ClinVar and >93% on ProteinGym DMS relative to VESM-3B), enabling high-throughput VEP under limited compute.
11. Clinical benchmark (ProteinGym ClinVar, 2,227 genes): sequence-only VESM models outperform other sequence-only PLMs (including ESM-C) and compete with or surpass methods using MSA/structure/population priors. VESM-3B shows balanced ROC behavior across specificity and sensitivity regimes.
12. AlphaMissense comparison: VESM-3B performance is stable across allele-frequency strata, while AlphaMissense shows strong dependence on MAF (consistent with circularity risks when population frequency informs clinical labels). After excluding variants overlapping AlphaMissense training (gnomAD v2 MAF > 1e-5), all VESM sizes outperform AlphaMissense on AUC and multiple calibrated metrics.
13. Modular use of structure: rather than retraining a joint model, they fine-tune the sequence component of ESM3 using VESM-style sequence-based loss to create VESM3, and combine VESM3 with VESM-3B into a structure-aware ensemble (VESM++). This improves performance on structure-dependent DMS assays (binding/stability/expression) while maintaining strong fitness/activity performance.
14. Cross-domain generalization: despite co-distillation being trained on human proteins, gains transfer strongly to nonhuman DMS assays, with disproportionately large improvements reported for viral proteins—even though ESM3’s released training data excluded viral sequences.
15. Beyond binary pathogenicity: using UK Biobank/Genebass summary statistics for 332 gene–phenotype pairs (blood biochemistry biomarkers), variant-level VESM scores correlate with single-variant effect sizes (β). VESM++ and VESM-3B yield the strongest gene–trait association signals across tested models.
16. Notably, VESM-3B recovers the correct pLoF direction of effect in 98.8% of significant gene–phenotype pairs and identifies many associations not detected by missense burden tests, suggesting utility for quantitative trait interpretation from summary statistics.
📜Paper: doi.org/10.1038/s41592…
#ProteinLanguageModels #VariantEffectPrediction #ComputationalBiology #HumanGenetics #ESM #ClinVar #ProteinGym #DeepMutationalScanning #UKBiobank #MachineLearning
English