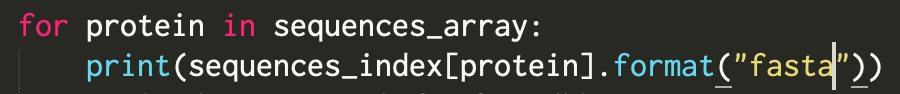
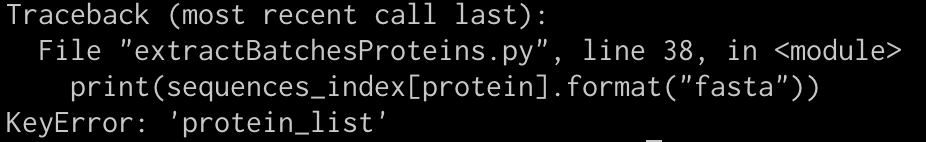
We don't have a dedicated Mastodon account, for now keep an eye on @OpenBio" target="_blank" rel="nofollow noopener">genomic.social/@OpenBio and #Biopython
English
Biopython Project
837 posts
@Biopython
Open-source bioinformatics libraries in Python



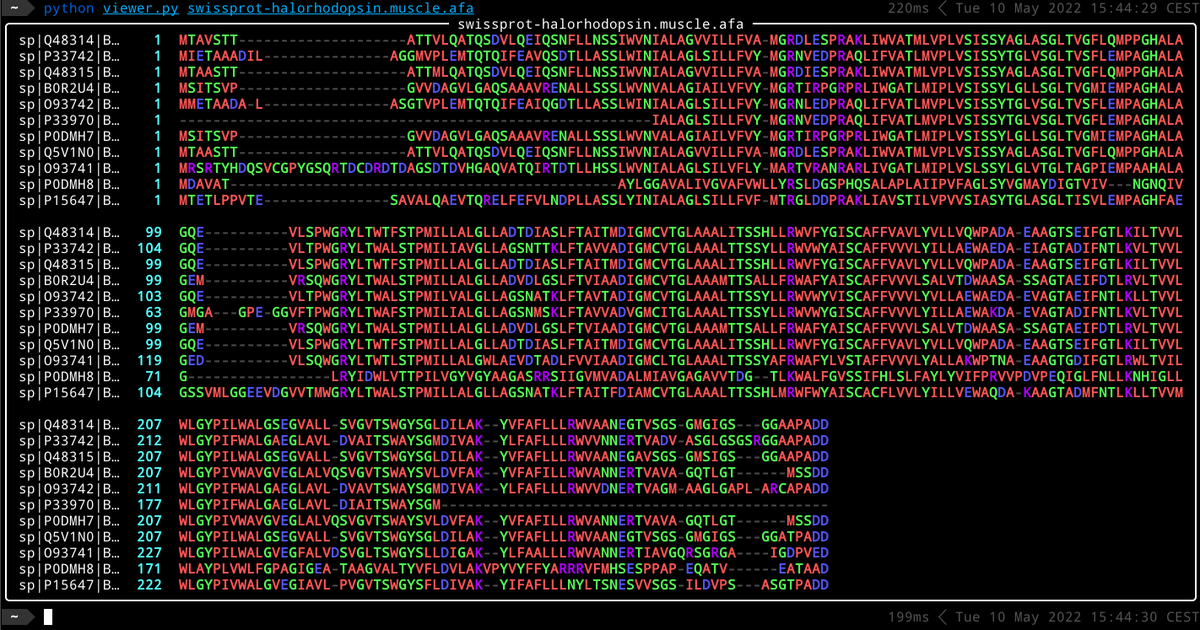





Any #Biopython @python friends out there who can write a tutorial on how to use this module? biopython.org/docs/dev/api/B… Trying to do this in Jupyter. What I need is an actual worked example.