🏆 Best Poster Award
ESMRank learns and integrates mutational constraint from >2M variants to predict variant effects directly from protein sequence.
📰: shorturl.at/bK8HL
Great and inspiring science at the AI & Biology Conference in Heidelberg - 🙏🏻 @EMBLEvents#EESAIBio
Work on variant effect prediction or protein engineering?
ESMRank: a learning-to-rank model trained on >1M variants from overlapping MAVE datasets reveals a transferable axis of mutational constraint.
Preprint: biorxiv.org/content/10.648…#pLLMs#MaveDB#ProteinGym#Bioinformatics
PhD positions in my lab (AI for Science), but with special preference for people who complement our current activities or are enthusiastic about contributing to our on going work. Looking for technically strong, independent and proactive candidates. chalmers.se/en/about-chalm…
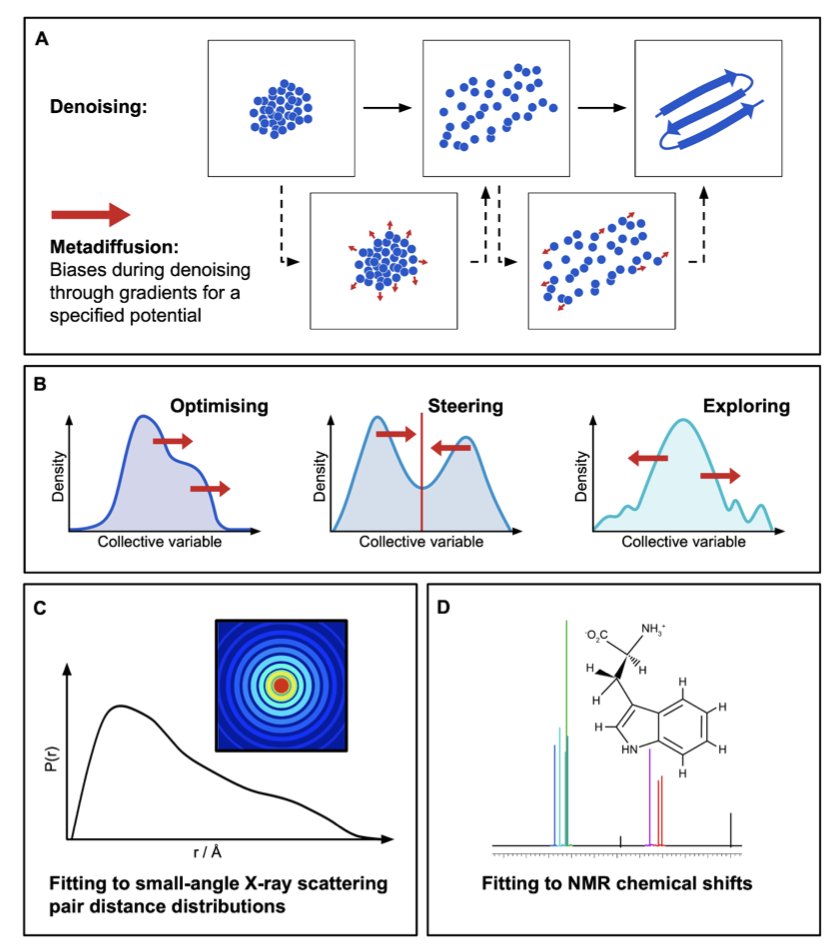
Metadiffusion: Inference-Time Meta-Energy Biasing of Biomolecular Diffusion Models
1. The authors introduce metadiffusion, a method that adds an inference-time meta-energy biasing layer on top of pretrained biomolecular diffusion models like Boltz-2, enabling diverse conformational ensemble generation without any retraining or fine-tuning.
2. The approach supports three complementary modes: optimisation (pushing collective variables to extrema), steering (targeting specific values), and exploration (maximizing inter-sample diversity through Gaussian repulsion potentials).
3. Metadiffusion generates conformational ensembles whose residue-level flexibility patterns closely match molecular dynamics simulations, achieving mean Pearson's R of 0.81 with ATLAS MD data—approaching the reproducibility ceiling between independent MD runs themselves.
4. The method enables controlled exploration of collective variables including radius of gyration, hinge angles, solvent-accessible surface area, and pairwise RMSD, allowing researchers to traverse alternative binding poses across proteins, nucleic acids, and ligands.
5. Through gradient-guided denoising, metadiffusion can steer ensembles to match experimental observables including SAXS pair distance distributions and NMR chemical shifts, as demonstrated with class V GTP aptamer and human calmodulin.
6. The technique operates orders of magnitude faster than microsecond-scale MD simulations while maintaining physical plausibility, with generated structures readily improvable through energy minimization to resolve occasional bond breaks and steric clashes.
7. As a model-agnostic framework, metadiffusion can theoretically extend to other diffusion-based structure predictors including AlphaFold3, OpenFold, and Chai-1, bridging the gap between single-structure prediction and ensemble-level experimental constraints.
💻Code: github.com/metadiffusion/…
📜Paper: biorxiv.org/content/10.648…#compbiol#structuralbiology#proteindynamics#diffusionmodels#machinelearning#alphafold#boltz2#conformationalensemble#SAXS#NMR#metadynamics#biomolecularsimulation
Today we share a technical report demonstrating how our drug design engine achieves a step-change in accuracy for predicting biomolecular structures, more than doubling the performance of AlphaFold 3 on key benchmarks and unlocking rational drug design even for examples it has never seen before.
Head to the comments to read our blog.
@PriyandarshanK The MSA is essentially the experimental data used as input to AlphaFold. AF didn't solve the protein folding problem, it solved the graph extraction (from MSA) and 3D embedding problem.
Knowing the MSA, lets one analyze if low confidence prediction is simply due to lack of data.
Excited to be at #EBSA2025!
Today I'll present my postdoc project carried out under the supervision of Lucie Delemotte and with the collaboration of @SaraILiin and her amazing team!
What if we could universally recombine, insert, delete, or invert any two pieces of DNA?
In back-to-back @Nature papers, we report the discovery of bridge RNAs and 3 atomic structures of the first natural RNA-guided recombinase - a new mechanism for programmable genome design
A huge milestone for personalised #mRNA therapeutics for inherited disease! A baby with an ultra-rare mitochondrial disease was treated at UPenn with an mRNA-encoded gene-corrector. The mRNA was made to treat a single individual in just 7 months!
nejm.org/doi/full/10.10…
Our several-years-old fix to ProteinMPNN's tendency to make weird antibody CDR seqs is finally out. We run an antibody LM in parallel & added its logits to ProteinMPNN's, fixing most issues we encountered. It also increased % of HER2-binding trastuzumab designs >10-fold
📢Ritorna la Walk of Life di Torino!
La ricerca corre, corri anche tu: 🏃♀️🏃♂️il 25 maggio ti aspettiamo alla III edizione della Walk of Life di Torino, un’occasione di incontro per dare una speranza concreta a chi affronta una malattia genetica rara.
bit.ly/4j5MW7U
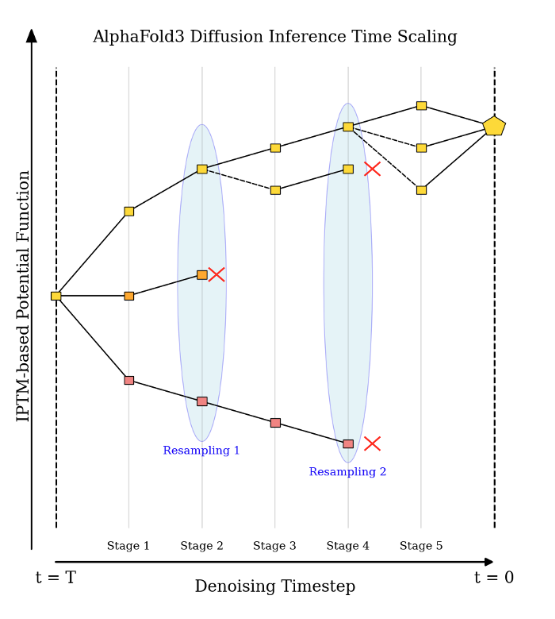
Protein modeling may be swiftly pivoting from one-shot prediction to steerable, context-aware generation.
Example: FKSFold uses Feynman-Kac control to inject ipTM rewards into AlphaFold3’s diffusion and rescues 3 of 8 tough molecular-glue ternaries.
A Perspective by Stephanie Wankowicz and James Fraser discusses ways macromolecules use conformational entropy to control binding, catalysis, and allostery
nature.com/articles/s4158…