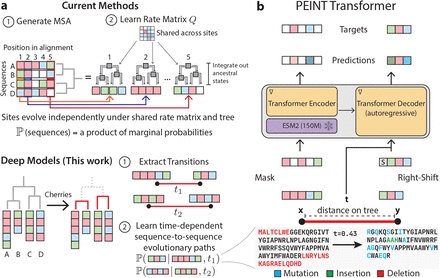
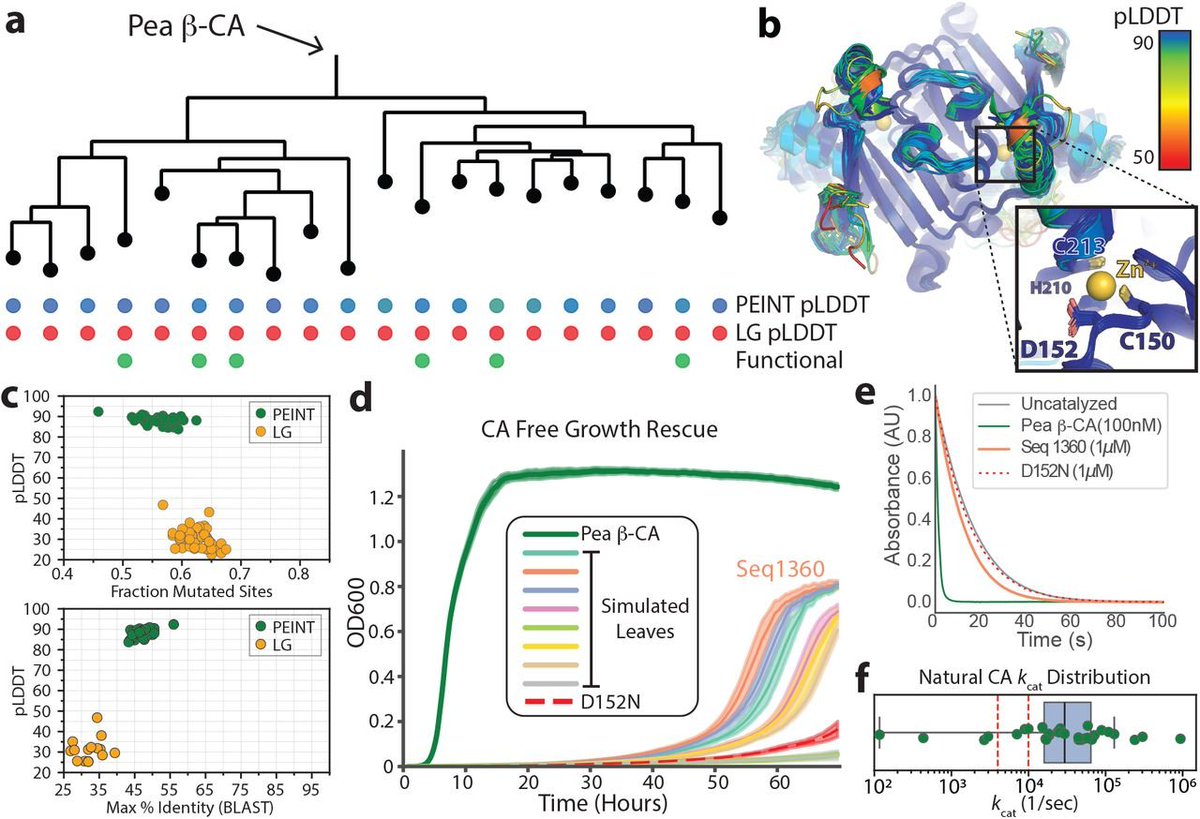
TechBio Transformers 已转推

🧬 Check out this new tech blog on designing protein binders by generative search in Proteina-Complexa:
developer.nvidia.com/blog/designing…
English
TechBio Transformers
642 posts


@TechBi0
Global Bio x AI Community. A third place for scientists, product managers, computational biologists, engineers and academics working in AI & Software for Bio



Mistral AI just released a text-to-speech model, which it says beats ElevenLabs And the weights are free!

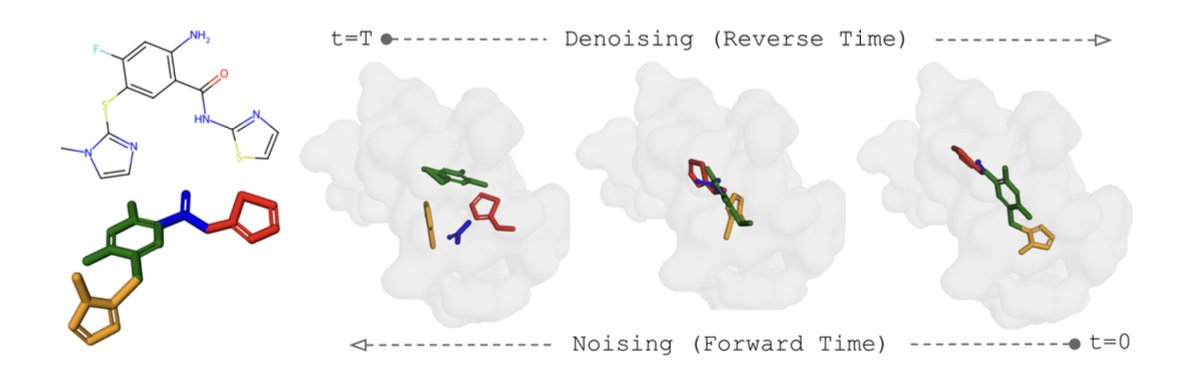
Chemical biologist Derek Lowe wrote about Boltz-2 in his column this week & a recent paper evaluating the AI/ML model for cofolding ligands & proteins. His column: "AI-Predicting Compound Affinity. We Aren't There Yet." He also brought up AlphaFold 3 docking predictions. A link to the column & the recent paper is in the reply







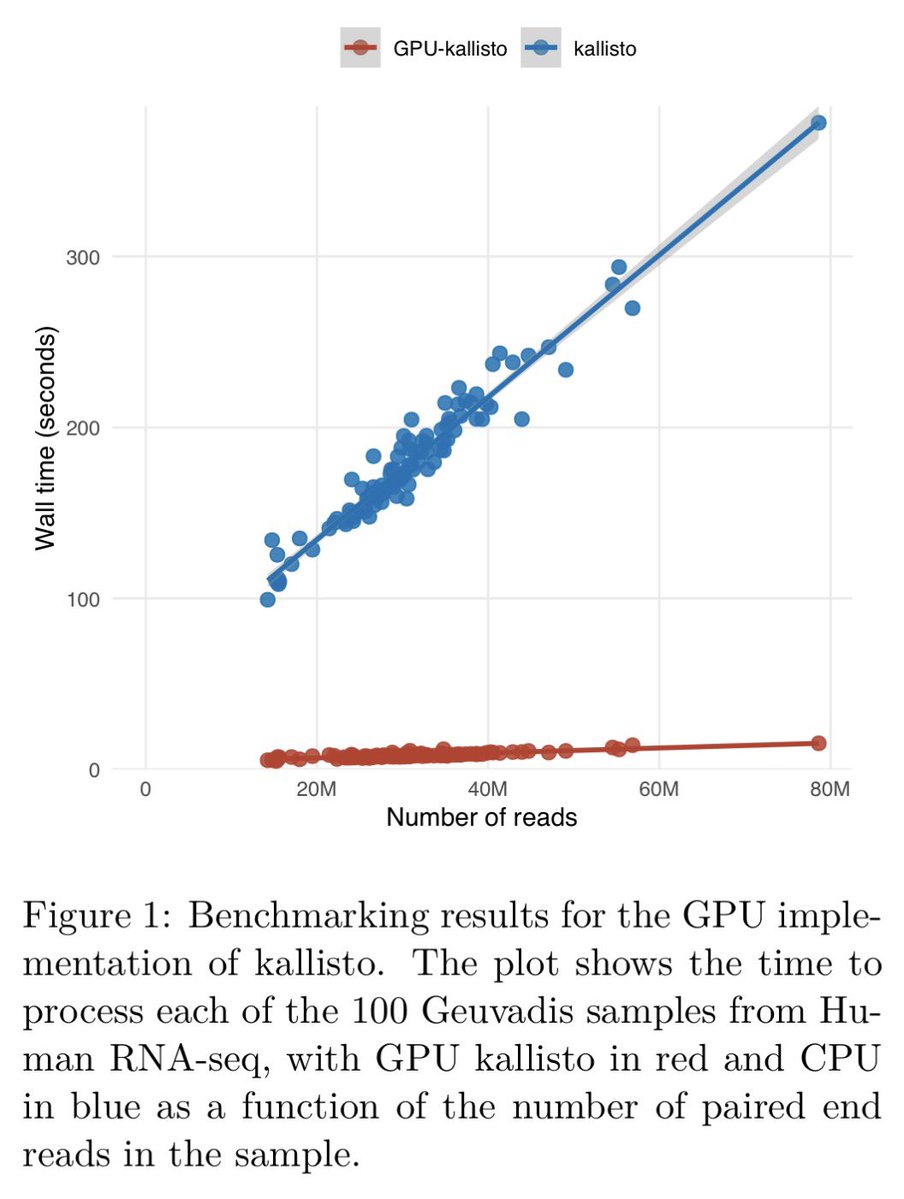
RNA-seq analysis in seconds using GPUs | bioRxiv biorxiv.org/content/10.648…


Nano Banana 2 is our new faster and better SOTA image generation & editing model! It uses Gemini’s amazing world understanding + grabs real-time info w/ search to create higher quality outputs. Available in @GeminiApp, @GoogleAIStudio, @FlowbyGoogle, Search & Vertex - enjoy!