
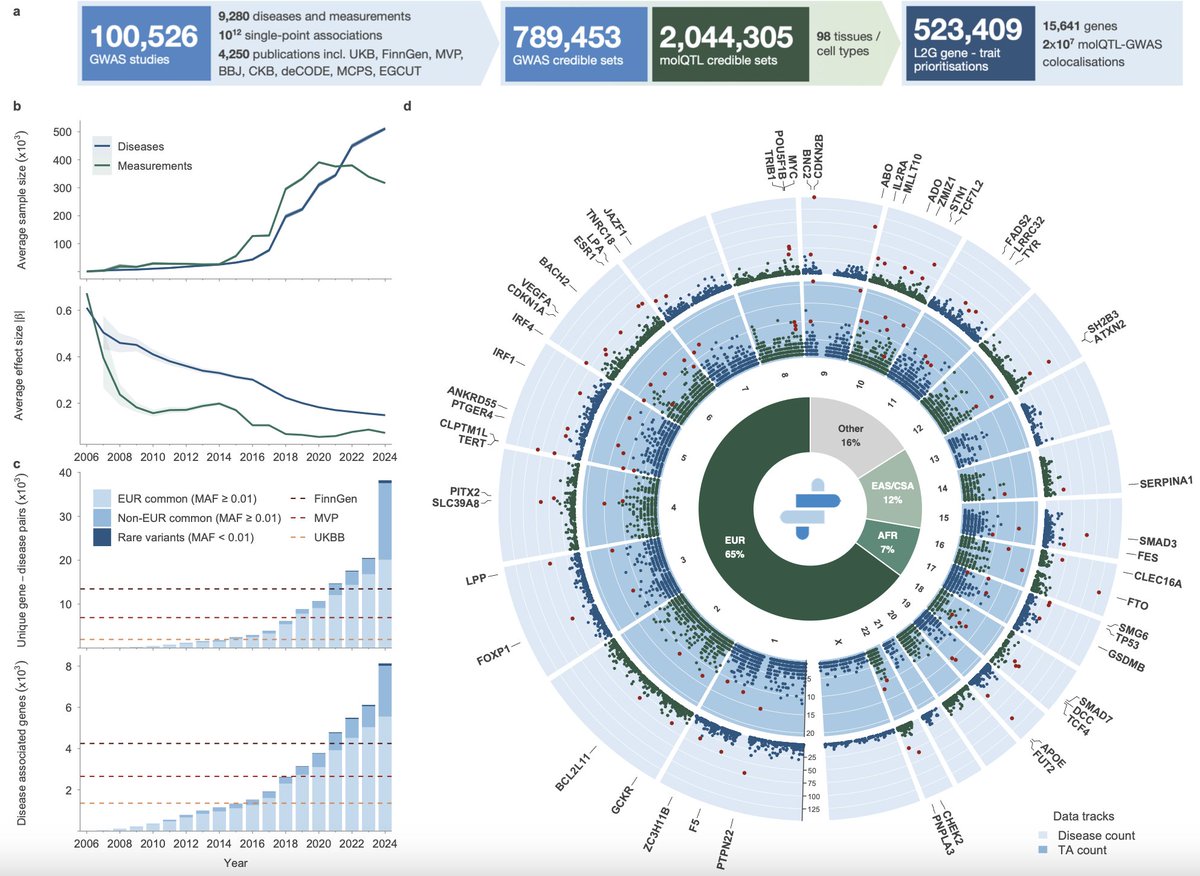
🧬New massive study of more than one trillion variant–trait associations suggests that drug-target success may lie in the middle ground between low and high pleiotropy. 💊
The authors show that most genes are pleiotropic — meaning that the same gene or genetic variant can be associated with multiple traits or diseases. Among 8,285 disease-associated genes, about 64% were linked to more than one disease. Moreover, 4,743 genes were associated with multiple therapeutic areas.
This is highly important for drug discovery, because a therapeutic target is usually embedded in a network of molecular and biological mechanisms rather than linked to a single isolated outcome.
As expected, in most cases the effects of pleiotropic lead variants were concordant: 92.5% showed the same direction of association across diseases. In other words, the genetic variant tended to increase the risk of the associated diseases.
But there were also opposite examples. For instance, the APOE p.Cys130Arg variant was associated with lower risk of age-related macular degeneration and non-alcoholic fatty liver disease, but higher risk of Alzheimer’s disease. This illustrates why such studies are especially important: even before clinical trials, we can start to account for potential safety liabilities.
Overall GWAS support was associated with a higher chance of clinical success — roughly a threefold increase.
Perhaps the most important result is that the association between pleiotropy and drug-target success is non-linear. Too little pleiotropy may indicate weak or narrow biological involvement. Too much pleiotropy may imply systemic effects and safety liabilities. The optimum appears to be a target with a strong enough biological signal, but without an excessively broad phenotypic footprint.
Proud of my former colleague and co-author of this paper, @tskir1 — congratulations!
platform.opentargets.org
biorxiv.org/content/10.648…
#GWAS #OpenTargets #Pleiotropy #DrugDesign #ComplexDiseases
English