
Andrew MacBride
5K posts

Andrew MacBride
@swimsf
biology + computers + a leavening of snark | 👨👨👧👧🏳️🌈| cancer genomics 🧬 | ML | biomaterials | startups | @Cal 🐻 @Stanford @UniOfOxford @OxfordNano
iPhone: 37.763271,-122.449059 가입일 Nisan 2009
5.9K 팔로잉989 팔로워

It's coming! I'm cumming!
Pop Base@PopBase
Madonna unveils logo for her new album ‘Confessions II.’
English
Andrew MacBride 리트윗함

What if AI could invent enzymes that nature hasn’t seen? 👩🔬🧑🔬
Introducing 🪩 DISCO: Diffusion for Sequence-structure CO-design
14 rounds of directed evolution and over a year of wet lab work. That's what it took to engineer an enzyme for selective C(sp³)–H insertion, one of the most challenging transformations in organic chemistry.
DISCO surpasses this with a single plate. No pre-specified catalytic residues, no template, no theozyme, no inverse folding, just joint diffusion over protein sequence and structure.
📝 Blog: disco-design.github.io
📄 Paper: arxiv.org/abs/2604.05181
💻 Code: github.com/DISCO-design/D…
English
Andrew MacBride 리트윗함


Andrew MacBride 리트윗함

Start a company in AI for Science.
The Encode: AI for Science fellowship offers a year of freedom to build what matters -- salary, 100k GBP of compute, and partnership with the top scientists in the UK.
No equity or fees, it's a fully funded fellowship!
Apply by March 28

English
Andrew MacBride 리트윗함

Software horror: litellm PyPI supply chain attack.
Simple `pip install litellm` was enough to exfiltrate SSH keys, AWS/GCP/Azure creds, Kubernetes configs, git credentials, env vars (all your API keys), shell history, crypto wallets, SSL private keys, CI/CD secrets, database passwords.
LiteLLM itself has 97 million downloads per month which is already terrible, but much worse, the contagion spreads to any project that depends on litellm. For example, if you did `pip install dspy` (which depended on litellm>=1.64.0), you'd also be pwnd. Same for any other large project that depended on litellm.
Afaict the poisoned version was up for only less than ~1 hour. The attack had a bug which led to its discovery - Callum McMahon was using an MCP plugin inside Cursor that pulled in litellm as a transitive dependency. When litellm 1.82.8 installed, their machine ran out of RAM and crashed. So if the attacker didn't vibe code this attack it could have been undetected for many days or weeks.
Supply chain attacks like this are basically the scariest thing imaginable in modern software. Every time you install any depedency you could be pulling in a poisoned package anywhere deep inside its entire depedency tree. This is especially risky with large projects that might have lots and lots of dependencies. The credentials that do get stolen in each attack can then be used to take over more accounts and compromise more packages.
Classical software engineering would have you believe that dependencies are good (we're building pyramids from bricks), but imo this has to be re-evaluated, and it's why I've been so growingly averse to them, preferring to use LLMs to "yoink" functionality when it's simple enough and possible.
Daniel Hnyk@hnykda
LiteLLM HAS BEEN COMPROMISED, DO NOT UPDATE. We just discovered that LiteLLM pypi release 1.82.8. It has been compromised, it contains litellm_init.pth with base64 encoded instructions to send all the credentials it can find to remote server + self-replicate. link below
English
Andrew MacBride 리트윗함

just got invited to peer review a paper I'm one of the authors on
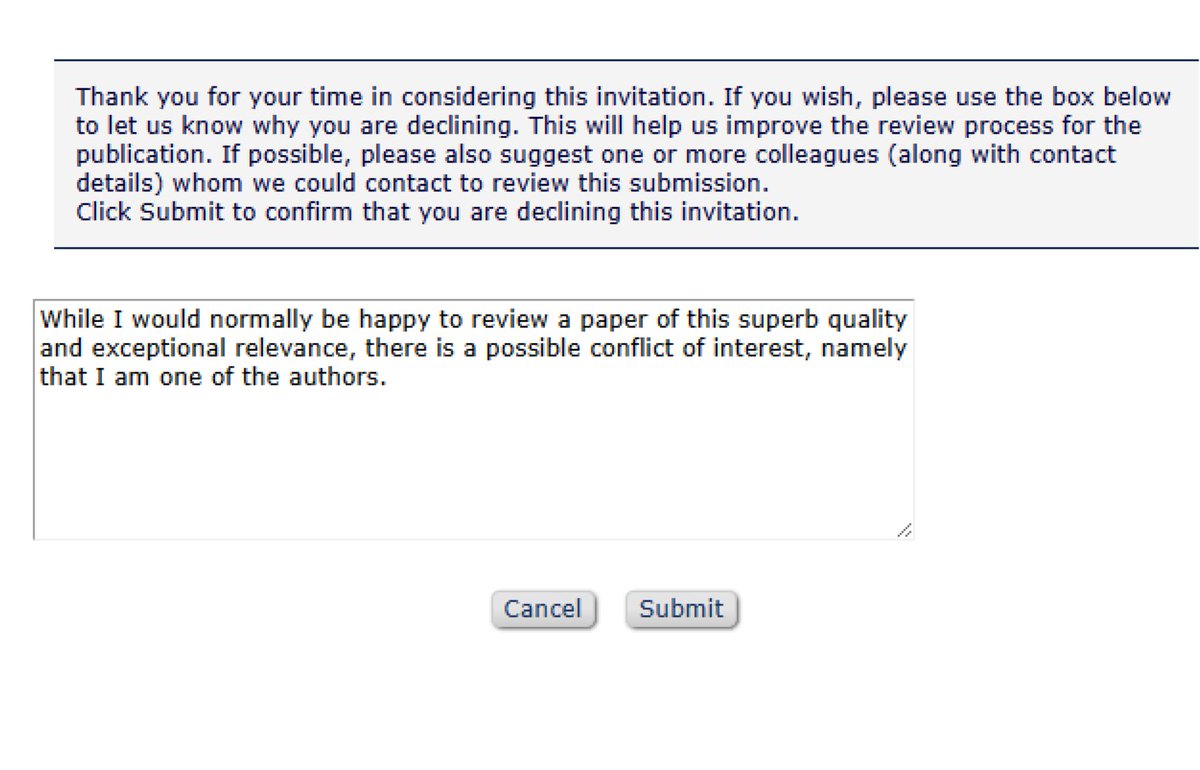
English
@AdamJSchwarz @morgfair She is all of us right now.
(well, the non-crazy ones at least)
English

Japanese PM Sanae Takaichi's reaction as Trump says "Who knows better about surprise than Japan? Why didn't you tell me about Pearl Habour?"
Undoubtedly the worst American diplomatic gaffe in post-war US-Japan history.
English
Reporter: Why didn't you notify Japan that you were going to attack Iran?
Trump next to the Japanese PM:
"Who knows better about surprise than Japan? Why didn't you tell me about Pearl Habour? You believe in surprise I think much more so than us."
English



@NateKrefman @phylogenomics This is definitely a case of someone not knowing what they don’t know.
AI for bio is absolutely revolutionary; it’s also absolutely not close to finished.
English


Andrew MacBride 리트윗함

People will be like "I can't believe they made star trek woke" and then you tune into 90s star trek and there's a transgender worm walking across the transgender carpet in the gay communist space station

English
OKAY thank you everyone. I’m going to say so that I don’t have a panic attack taking on too much that I’m going to close this current batch, but if you’re interested I’d love to put together a waitlist 🫣🫣🫣
🌟 Tyler Koberstein 🏳️🌈@t_kobs
✨LET ME DRAW YOU!✨ To offset my layoff, I’m opening up commissions! I’d love if I could draw you. Prices are on this sheet. DM me for more information. If you can’t afford it right now, I’d greatly appreciate just spreading the word. ❤️❤️❤️
English
Sad news time: after 6+ years, this is my last week employed at Nickelodeon.
English
@t_kobs Oh no! This short is of course brilliant, which makes it even sadder. 🫂
English
Andrew MacBride 리트윗함

Modeling all 28,000 genes at once: a foundation model for single-cell transcriptomics
Every cell in your body carries the same genome, yet a neuron looks and behaves nothing like a liver cell. The difference lies in which genes are turned on or off—and at what level. Single-cell RNA sequencing (scRNA-seq) lets us measure that expression profile one cell at a time, revealing rare cell populations, gene regulation, and drug response at unprecedented resolution.
Foundation models pretrained on millions of cells have become powerful tools for analyzing these data. But they all share a practical compromise: restricting their attention mechanism to ~2,000 highly expressed genes and discarding the remaining ~26,000. Many of those excluded genes, despite low expression, act as regulatory switches, fine-tuners of signaling pathways, and drivers of context-specific responses like immune activation or drug resistance. Ignoring them means learning an incomplete picture of the cell.
Ding Bai and coauthors address this with scLong, a billion-parameter model pretrained on 48 million cells that performs self-attention across all 27,874 human genes. To make this feasible, they use a dual encoder: a large Performer (42 layers) processes the top 4,096 high-expression genes, while a smaller one (2 layers) handles the remaining ~24,000. Both outputs merge through a full-length encoder capturing cross-group interactions. scLong also integrates Gene Ontology knowledge via a graph convolutional network, embedding each gene with information about its known functions, processes, and cellular localization—context that expression data alone cannot provide.
Results are consistent and broad. In predicting transcriptional responses to genetic perturbations, scLong achieves a Pearson correlation of 0.63 on unseen perturbations, compared to 0.56–0.58 for existing models and GEARS. It outperforms Geneformer, scGPT, and DeepCE on chemical perturbation prediction across all metrics, reaches 0.873 Pearson for cancer drug response, and surpasses both Geneformer and DeepSEM in gene regulatory network inference.
The broader point: in biological foundation models, what you choose to attend to shapes what you can learn. By including low-expression genes and grounding representations in functional knowledge, scLong shows that scaling context—not just parameters—is key to capturing the full complexity of cellular regulation. A principle relevant wherever long-range feature dependencies are biologically meaningful but computationally expensive to model.
Paper: nature.com/articles/s4146…

English
@rossiadam It really was. Getting to interact with very senior engineers was an amazing education; I was there for four years and it felt like another degree.
English

In 2004 the Sun Fire E25k was the apex predator of computing. This was the largest single-image Unix machine that money could buy ($2M fully loaded).
This bad boy had:
• 36 UltraSPARC IV processors (72 cores)
• 576 GB RAM
• 18 fully hot swappable CPU/memory boards
• gigabit ethernet and fibre channel HBAs
• Redundant power supplies, cooling zones, draw 15kW of power
I deployed software to these things. Uptime was measured in years. This was THE ONE.
Man I miss Sun.

English
@cardon_brian I swear to God, that is the gayest thing I’ve ever seen.
English

I’m tired of MAGA saying that they’re Alpha males. Look at Gunther Eagleman chilling at a Starbucks showing the world what an impotent, Low T individual looks like. Who the fuck eats a banana like this? I’m going to make a piece of Avocado toast.
English
