
Daniel Portik
852 posts

Daniel Portik
@DPortik
Bioinformatics scientist @PacBio. I work on metagenomics, methylation, and assembly, but also enjoy phylogenomics and frog/lizard evolution. 🦠 🐸 🦎 🧬
Katılım Şubat 2021
3.4K Takip Edilen2.4K Takipçiler



Whoa! @nanopore was certainly quiet about axing ElysION after a year or so and a bit over a dozen placed
aseq.substack.com/p/oxford-fy202…
English
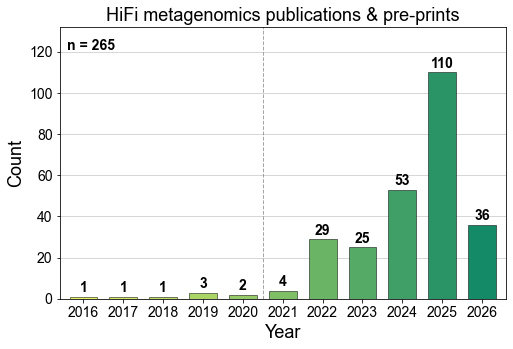
We've seen a big increase in the number of long-read #metagenomics publications, especially those using #PacBio HiFi data. Also very encouraging to all the new tools for long-read metagenome assembly & taxonomic profiling.
Complete list of publications: github.com/PacificBioscie…
English
Another #metagenome assembly algorithm for #PacBio HiFi data - introducing Alice!
I'll be very interested to see how this performs on some of our trickier datasets.
Happy to see continual development of tools for long-read #metagenomics.
doi.org/10.1101/2025.0…
English
Daniel Portik retweetledi

Complex structural variant visualization with SVTopo. #GeneticVariants #StructuralVariants #VariantsVisualization #BMCgenomics
bmcgenomics.biomedcentral.com/articles/10.11…

English
Daniel Portik retweetledi
PacBio: $300 Genome Via Chemistry Update 🧬🖥️
omicsomics.blogspot.com/2025/10/pacbio…
#ASHG2025
English
New Banfield lab pre-print highlighting 1.5Mb megaplasmids found in human gut.
#Plasmid genomes were resolved using @PacBio HiFi sequencing and hifiasm-meta for #metagenome assembly. Host association was detected using #epigenetic signals.
doi.org/10.1101/2025.0…
English
This could be a neat method for species detection in #metagenomic datasets, but why on earth are we simulating PacBio and ONT error rates at >10%?
🫣🤔🫠
PacBio HiFi accuracy is >99% since 2019.
Very hard to interpret the results based on 90% accuracy.
biorxiv.org/content/10.110…
English
New #metagenome assembly comparison of PacBio, ONT, and Illumina in gut #microbiomes.
- "HiFi generated more cMAGs than ONT at every depth with either assembly method"
- "HiFi cMAGs had significantly higher completeness and lower contamination than ONT"
doi.org/10.1016/j.cell…
English
Daniel Portik retweetledi

Great keynote talk at #SFAF by Rob Knight, summarizing key innovations in #microbiome research over the past decade.
One recent highlight is how long reads have transformed #metagenome assembly, particularly @PacBio HiFi reads. The future is complete MAGs!

English
@BioMickWatson @kirk3gaard Totally agree on this.
Still nice to see some traction with proof of concept projects in academia, and more routine use for metagenome assembly and tool development.
As with most things, I feel like the academic wave is a prerequisite for industry adoption...
English
@kirk3gaard @DPortik As far as I can tell, this is an entirely academic wave. Illumina metagenomics is the absolute leader in commercial sequencing, biotech, and genomics
English
Do you think long-read #metagenomics with #PacBio #HiFi is gaining momentum?
I noticed there were more publications & pre-prints on this topic in 2024 (56) than 2022 + 2023 combined (54). Definitely encouraging!
Full pub list: github.com/PacificBioscie…

English
@ComptonSop92834 @nbcsandiego I took this video and you have my permission to use it.
English

@DPortik @nbcsandiego Hi! Hope you're safe. Did you take this video/photo? If so, can we have permission to use on Fox News Channel, Fox Business Network, Fox Nation, Fox Weather, Outkick, and all Fox News Edge affiliates across all platforms until further notice with courtesy to you?
English
#Fire in San Diego in Linda Vista/Fashion Valley area, now called #Friar Fire. I took this video from our apartment complex and hope this helps with the effort to extinguish it quickly.
We evacuated minutes after I recorded this. @nbcsandiego
English
@natfaitelson @nbcsandiego Yes this is a video I took myself, you have my permission to use it.
English

@DPortik @nbcsandiego Hi, reaching out from NBC7 San Diego. Did you film this video yourself? If so, will you give
NBCU permission to share it on all our platforms and partners?
English
@TavleenTarrant @nbcsandiego Yes this is my video, I took it myself. You have my permission to use it.
English

@DPortik @nbcsandiego Hi, I'm with NBC News. Did you take this yourself and if yes, can we have permission to use it across all NBCU partners and platforms? Thanks and stay safe
English
Daniel Portik retweetledi

A bright new day is here! 🦋 Follow us on Bluesky (bsky.app/profile/pacbio…) and stay connected for updates, innovations, and everything genomics. #PacBio

English
Another very insightful paper on #methylation and long-read sequencing by @gangfang_1, describing some important but frequently overlooked caveats.
Gang provides a fantastic summary in his post, but here's my take too😅:

Gang Fang@gangfang_1
Preprint alert 🚨 Cautions in the use of @nanopore sequencing to map DNA modifications: officially reported “accuracy” ≠ reliable mapping in real applications. We performed a critical assessment of nanopore sequencing (across different versions of models) for the detection of multiple forms of DNA modifications: while it is reliable for high-abundance modifications, including 5mC at CpG sites in human cells and 5hmC in human brain cells, it makes very high % false positive calls for low-abundance modifications, such as 5mC at CpH sites, 5hmC and 6mA in most human cell types [1]. Want to know why? Please read further (long): In 2022, we reported in @ScienceMagazine that 6mA is much less abundant in human cells than previously reported (mostly false positives) [2]. In 2023, we wrote a perspective in @NatureRevGenet to help navigate the common pitfalls for false positive in the mapping of DNA and RNA modifications [3]. With these two papers, we thought the importance of recognizing false positive calls has been adequately described. Unfortunately, this was not the case. New official models report nanopore seq can achieve >97.5% raw read accuracy in the detection of 5mC, 5hmC and 6mA, across all sequence contexts. New methods are to be released for mapping DNA damages, etc. Unfortunately, we do not think these “accuracies” are reliable in practical applications. To help biologists and methods developers, @kong_yimeng (first author of the 2022 & 2023 papers, now a PI) and I felt the need to do this study to critically assess nanopore seq for the detection of multiple forms of DNA mods. Thanks to team and collaborators: @KohliLab @ChristianLoo_ @XuesongRutgers. Why did we pick nanopore in this critical assessment? First of all, we have no bias among different sequencing platforms. In fact, we regularly use nanopore seq in our lab for several projects. However, we think there is a broad misunderstanding and over expectation that nanopore sequencing can map many forms of DNA modifications in any genome. A fundamental limitation is that the training and test data used in existing machine learning models do not consider the physiological levels of DNA modifications. Common pitfalls: 1. High accuracies in training and test data don't guarantee reliability in real applications. It depends on the abundance of DNA modifications (6mA is abundant in bacteria, but rare in human). 2. Reliability in one sequence context or genomic region doesn't ensure reliability in others. It depends on the abundance of DNA modifications at a specific sequence context or genomic region (5mC is abundant at CpG sites but not at CpH sites; 5mC is abundant on the nuclear genome but not on mtDNA). 3. Reliability in one cell type doesn't guarantee success in others. It depends on the abundance of DNA modifications in a specific cell type (5hmC is abundant in brain tissues but rare in blood). Why do each of these pitfalls involve the critical consideration of the abundance of DNA modification? This is because every genomic technology has a certain level of background noise in detecting DNA modifications. It will detect a certain level of DNA modification even when no modification was present in a negative control sample! Therefore, the method is only reliable if the physiological levels of DNA modification are sufficiently higher than background noise. How to navigate the pitfalls? To assist biologists and methods developers, we describe a framework for rigorous evaluation that highlights the use of false discovery rates (FDRs) along with rationally designed negative controls capturing both general background and confounding modifications. What is “confounding modification”? It turns out that modification-free negative controls are not enough. For example, the abundant 5mCpG in most human cell types can lead to increased levels of false positive 5hmC calls, much higher than the actual 5hmC levels in most human cell types (Fig. 1 attached) …… (1) This study highlights the urgent need to incorporate this framework in future methods development and biological studies. Using this framework, you can quantitatively evaluate the reliability of DNA modifications detected in your samples. (2) This study advocates prioritizing nanopore sequencing for mapping abundant, not rare, modifications in biomedical applications. Specifically, 5mCpG can be largely reliably mapped across mammalian cell types; 5hmCpG mapping by existing methods should be limited to tissues with high 5hmC abundance (e.g. brain tissues). In addition, our assessment does not support the use of nanopore sequencing to map endogenous* 6mA or various DNA damages in mammalian genomes due to their low abundance. (3) This framework is also applicable to RNA modifications. Although this work is focused on DNA modifications, the principles we outline—rigorous false positive recognition, use of negative controls, and FDR evaluation—are equally applicable to RNA modifications. [1] New preprint: biorxiv.org/content/10.110… [2] Critical assessment of DNA adenine methylation in eukaryotes using quantitative deconvolution science.org/doi/10.1126/sc… [3] Navigating the pitfalls of mapping DNA and RNA modifications nature.com/articles/s4157… [*] Please note the detection of exogeneous DNA modifications is different from mapping natural endogenous DNA modifications, because abundant exogeneous DNA modifications are added to the genome as in Fiber-seq, DiMeLo-seq, etc.
English