Jean-Philip Piquemal@jppiquem
Speaking today at the @isqbp 2024 conference about new important results that we obtained in #quantumcomputing for #compchem. Check out the new preprint:
"Shortcut to Chemically Accurate Quantum Computing via Density-based Basis-set Correction"
arxiv.org/abs/2405.11567
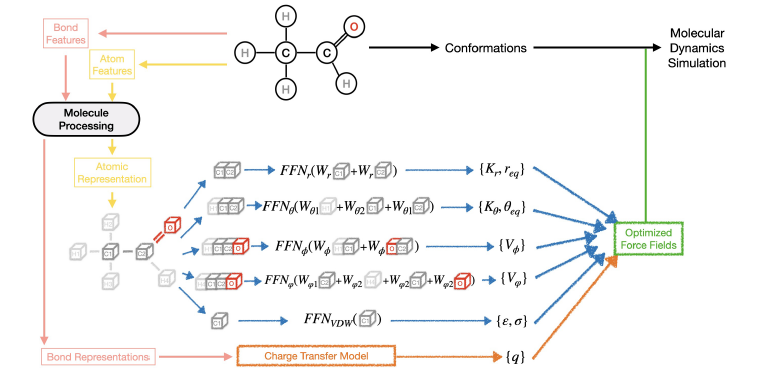
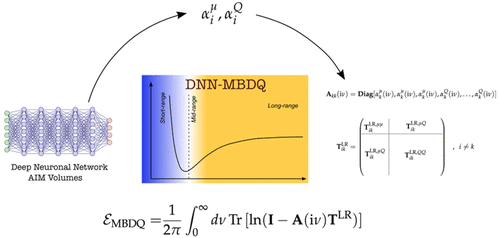
We present a new hybrid computing scheme for getting accurate energies and first-order properties of small molecules. The use of GPU-accelerated emulation allows us to obtain quantitative results at with ADAPT-VQE that would otherwise require brute-force quantum simulations using far more than 100 logical qubits. It is grounded on on-the-fly pivoted Cholesky-like adaptive basis sets (System-Adapted-Basis-Sets or SABS) and on a specific density basis set embedding of the quantum ansatz denoted density-based basis-set correction that allows recovering missing dynamical correlation effects due to basis set incompletess. Remaining missing basis effects can be included via a cheap Hartree Fock correction. We converge the ground-state energies of four systems (He, Be, H2, LiH) within chemical accuracy of the CBS full-configuration-interaction reference, while offering a systematic increase of accuracy beyond a double-zeta quality for various molecules up to the H8 hydrogen chain. We also obtain dissociation curves for H2 and LiH that reach the CBS limit whereas for the challenging simulation of the N2 triple-bond breaking, we achieve a near-triple-zeta quality at the cost of a minimal basis-set.
A nice feature of the approach is that it can be also used as "a posteriori" correction to any real QPU computation.
Stellar work by @Dr_DiataTraore @OlivierApp @ChemCesar I.-M. Lygatsika and great interdisciplinary work between chemistry, physics, #HPC and mathematics. Great collaboration between @LCT_UMR7616 (J. Toulouse, E. Giner), LJLL (Y. Maday), @qubit_pharma ( @alberto_peruzzo @EPosenitskiy) and @nvidia ( @khammernik). Funding from @ERC_Research (project EMC2) and PEPR Epiq @AgenceRecherche. Computer time @Genci_fr, @Scaleway_fr and NVIDIA.