
@fulop_dan Indeed, strandedness of the libraries does not (presently) affect alignments.
--soloStrand option is necessary for assigning reads to genes in the single-cell gene expression.
English
Alex Dobin
148 posts
@a_dobin
Director of Bioinformatics @ArcInstitute Formerly: ENCODE; PI@CSHL Developer of STAR.




STARsolo preprint is out on bioRxiv: biorxiv.org/content/10.110… STAR release 2.7.9a: github.com/alexdobin/STAR… The major new feature is quantification of multi-gene (multi-mapping) reads/UMIs, which are necessary to detect expression from overlapping genes and paralogs. 1/5

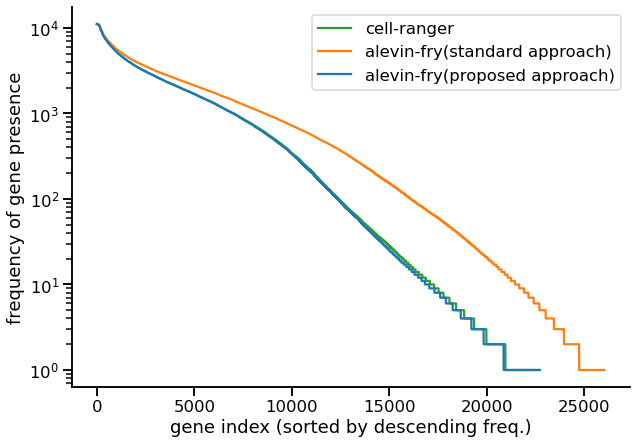
@hypercompetent @lpachter It’s getting late on the East coast, and still no blog from Lior, so I will make my presumptuous guess. I think Lior is trying to puzzle out why Kallisto to CellRanger correlation is lower in our Fig.4C biorxiv.org/content/10.110… vs. their Fig.2D nature.com/articles/s4158… 1/3






STARsolo preprint is out on bioRxiv: biorxiv.org/content/10.110… STAR release 2.7.9a: github.com/alexdobin/STAR… The major new feature is quantification of multi-gene (multi-mapping) reads/UMIs, which are necessary to detect expression from overlapping genes and paralogs. 1/5