Sabitlenmiş Tweet
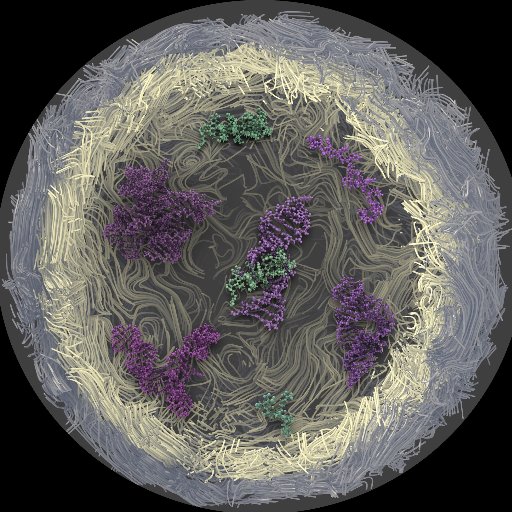
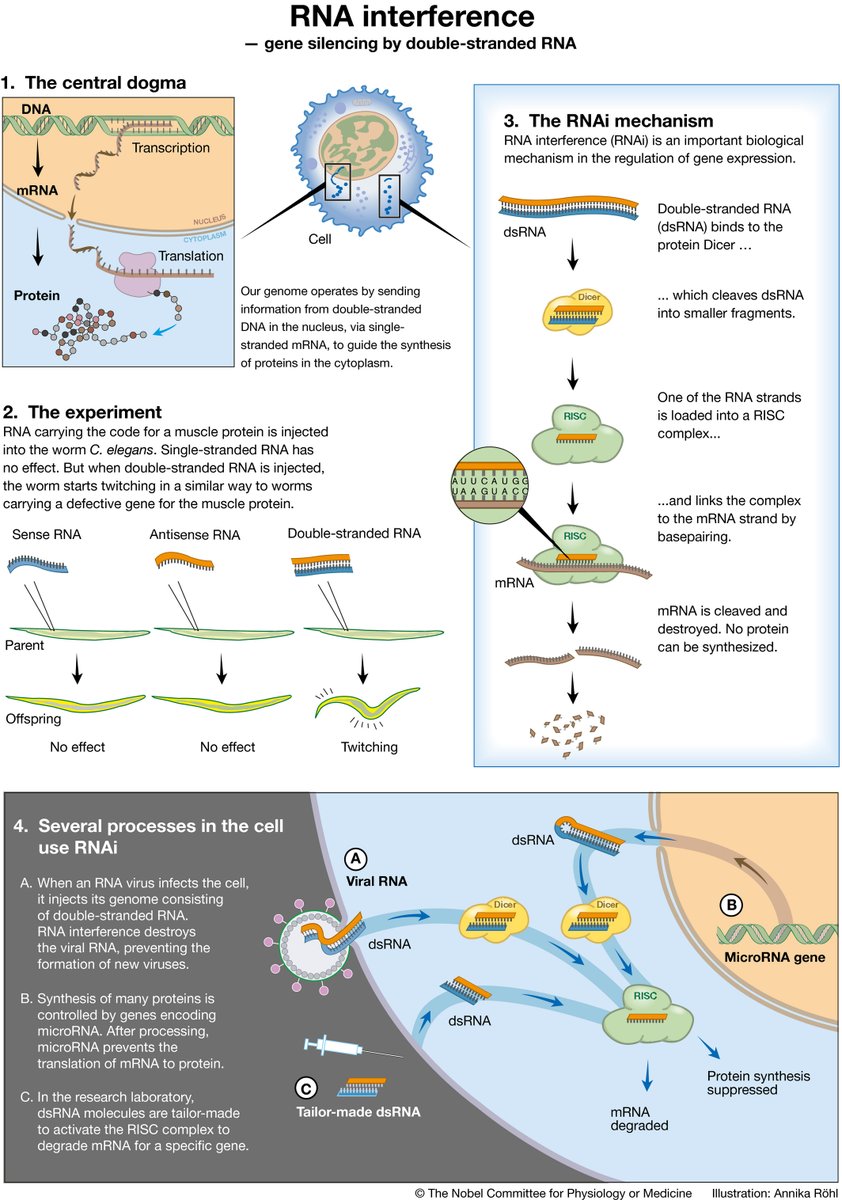
RNA to the rescue: RNA is one of the most promising targets for drug development given its wide variety of uses embopress.org/doi/full/10.15…
English
Fabrice Leclerc
193.3K posts
@rnomics
#RNomics, #RNA biology, RNA #bioinformatics, #RNA_World & #evolution, technologies & resources








⏰ Today is the LAST DAY for RNA Society members to submit entries for The RNA Day Art & Baking Contest! 🎨🧁 Thanks to @GenScript, our exclusive industry sponsor for this event, top entries will win cash prizes🏆#RNA #WorldRNAday #RNASociety #GenScript #RNASocietyRNADayContests