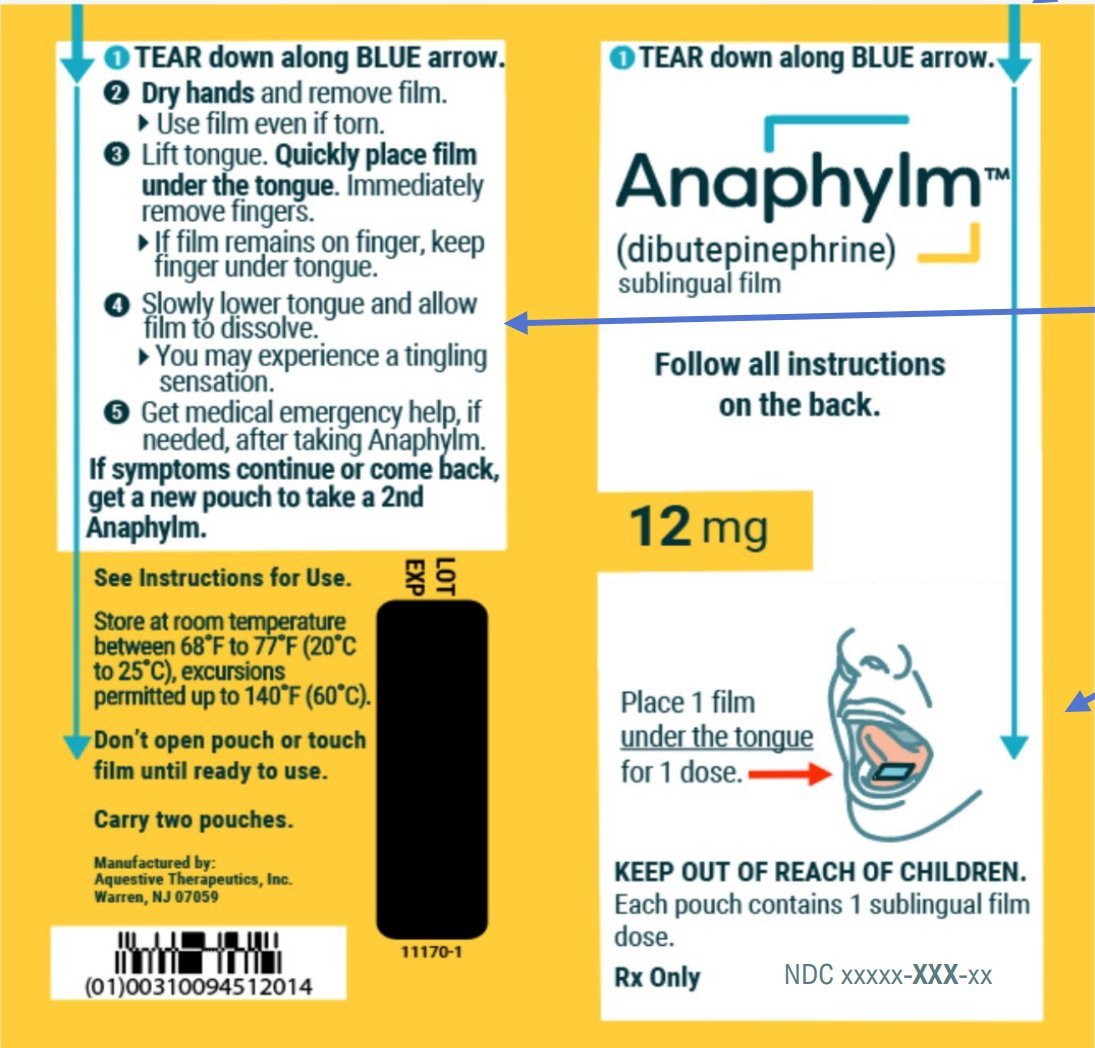
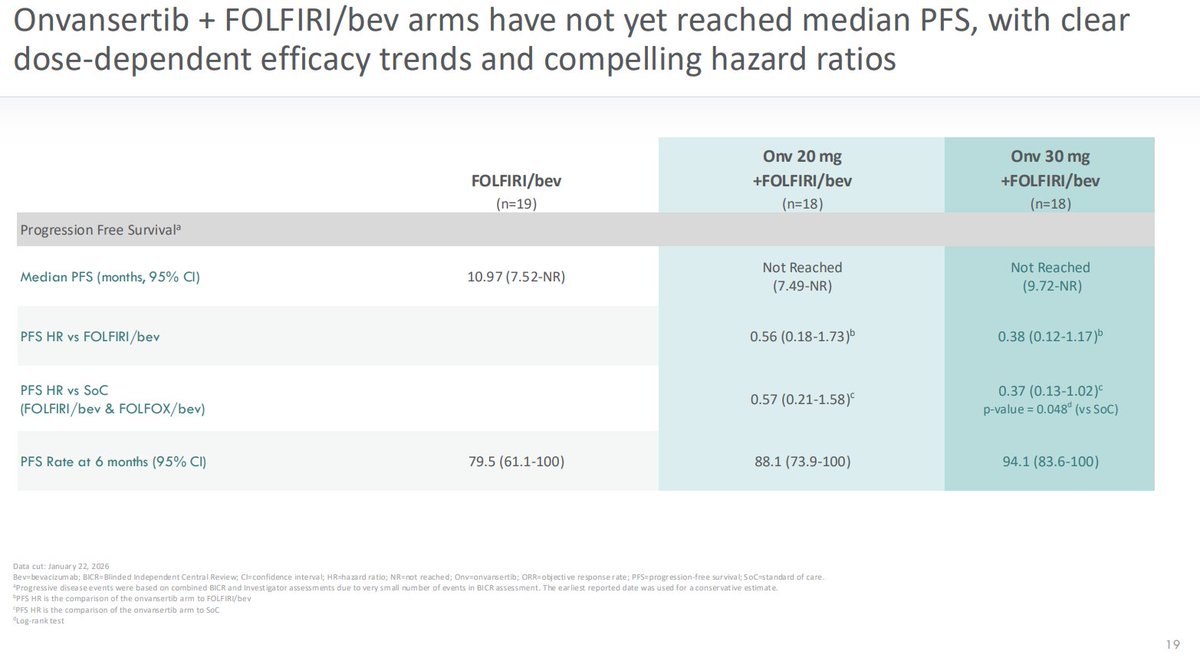
ทวีตที่ปักหมุด

I aspire to identify stocks that can be purchased now and held for a year to achieve long term capital gains. I seek to identify stocks that will at least double in value. I encourage you to consider these stocks yet do your own research before investing.
English