Carlos Acevedo-Rocha retweetledi

Compressing the collective knowledge of ESM into a single protein language model
Predicting whether a genetic variant is harmful or benign is one of the most consequential tasks in computational biology. A mutation in BRCA1 or PCSK9 can mean the difference between a healthy carrier and a serious disease. Most top-performing variant effect prediction (VEP) methods get their edge by combining protein language models (PLMs) with 3D structure, multiple sequence alignments (MSAs), or population genetics — extra information that is expensive, incomplete, or potentially circular in clinical settings.
Tuan Dinh and coauthors ask a sharp question: are sequence-only PLMs fundamentally limited, or just under-exploited?
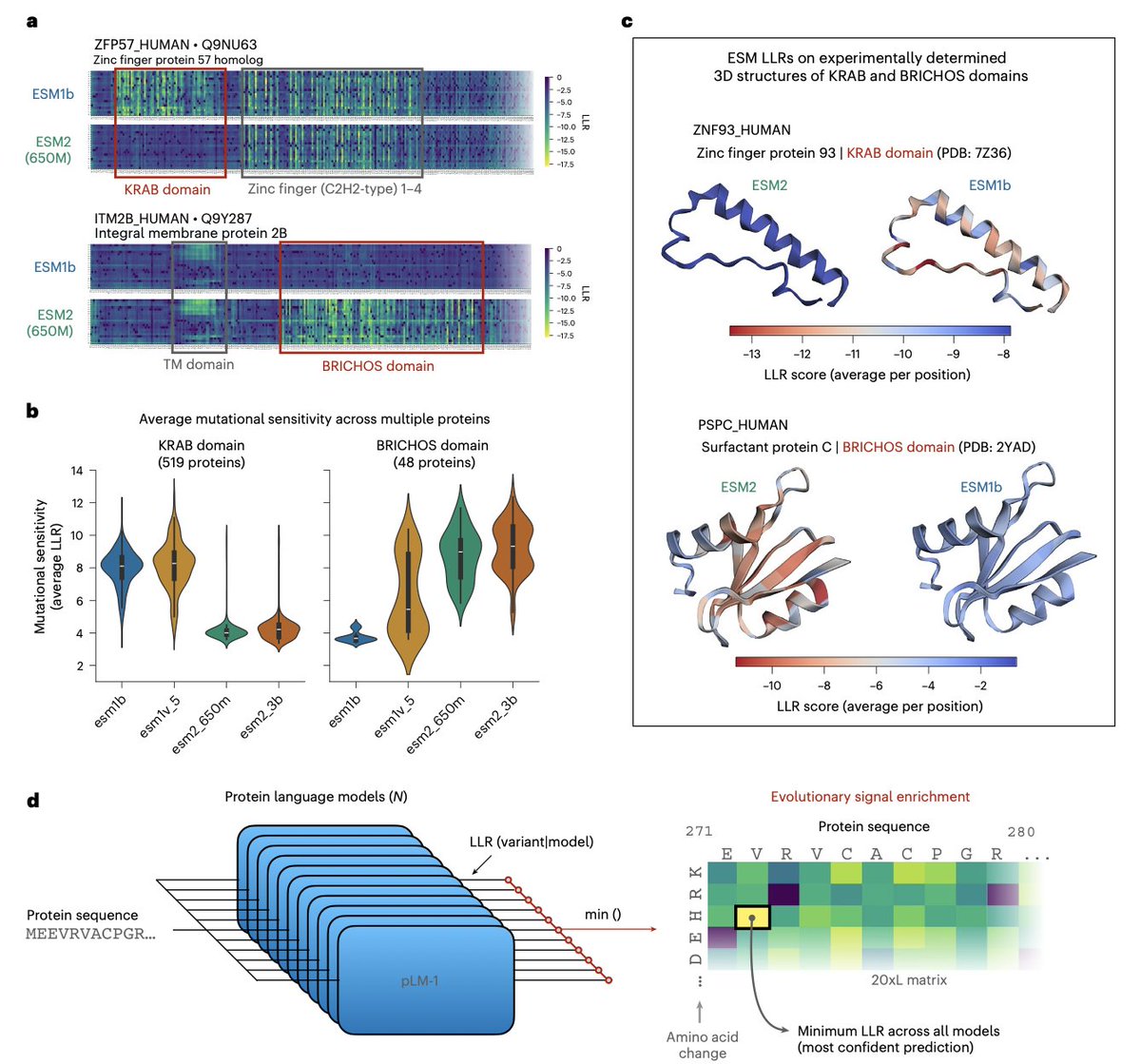
The key insight is that different ESM models — despite nearly identical architectures — have complementary blind spots. ESM2 reliably detects KRAB domains; ESM1b detects BRICHOS domains; neither catches what the other misses. Rather than averaging predictions (which dilutes rare signals), the authors select the minimum log-likelihood ratio across all models — the prediction most confident that a residue is mutationally sensitive. This signal then drives co-distillation of the entire ESM family into improved single models (VESM), through iterative rounds where models alternately teach and learn from each other.
The results are remarkable. VESM-3B, trained exclusively on unaligned sequences, matches or surpasses SaProt, PoET, TranceptEVE, and even AlphaMissense — a closed-source model trained on 3D structure, MSAs, and population allele frequencies. Critically, VESM maintains consistent performance across all allele frequencies, outperforming AlphaMissense precisely on the rare variants where clinical interpretation matters most. VESM scores also correlate quantitatively with continuous phenotypes in UK Biobank data, extending VEP from binary pathogenicity to quantitative trait prediction.
This is directly actionable: a sequence-only model at state-of-the-art accuracy removes hard dependencies on structural data or population databases, enabling scalable proteome-wide variant scoring — including for targets with no known structure, no deep alignment, and no prior clinical annotation.
Paper: Dinh et al., Nature Methods (2026) — CC BY 4.0 |
nature.com/articles/s4159…

English