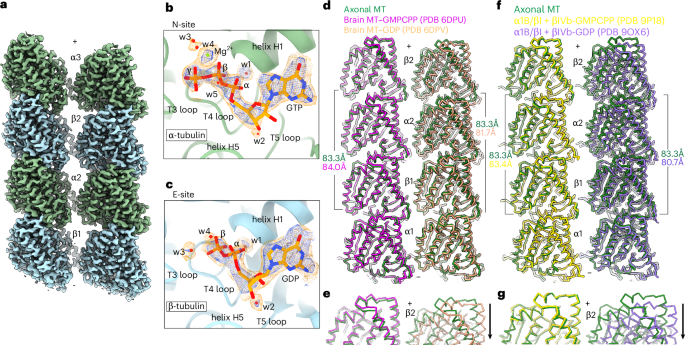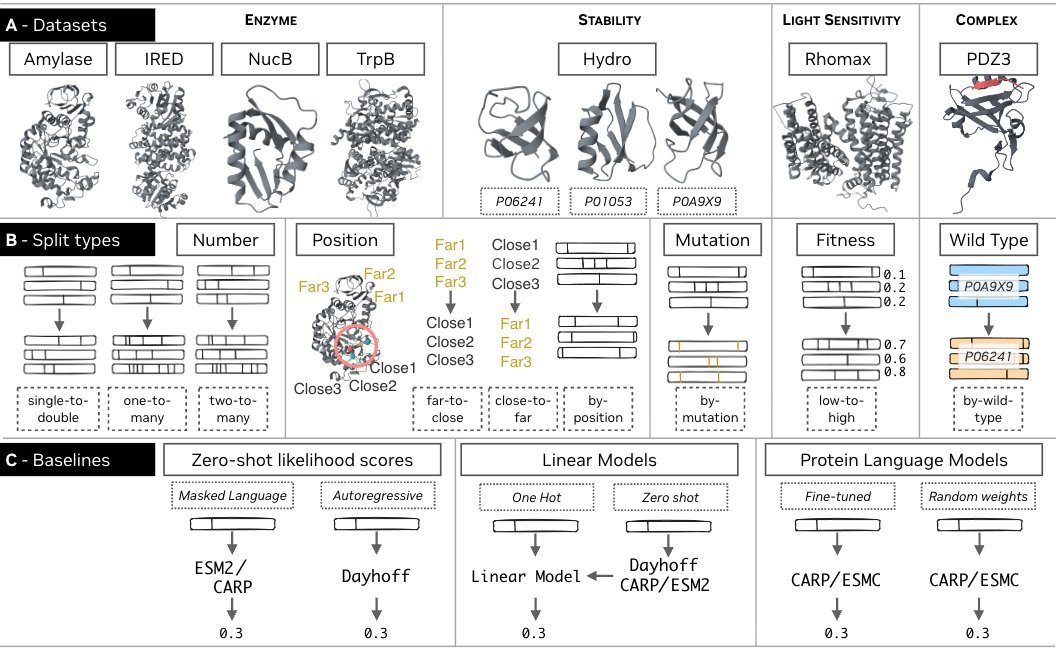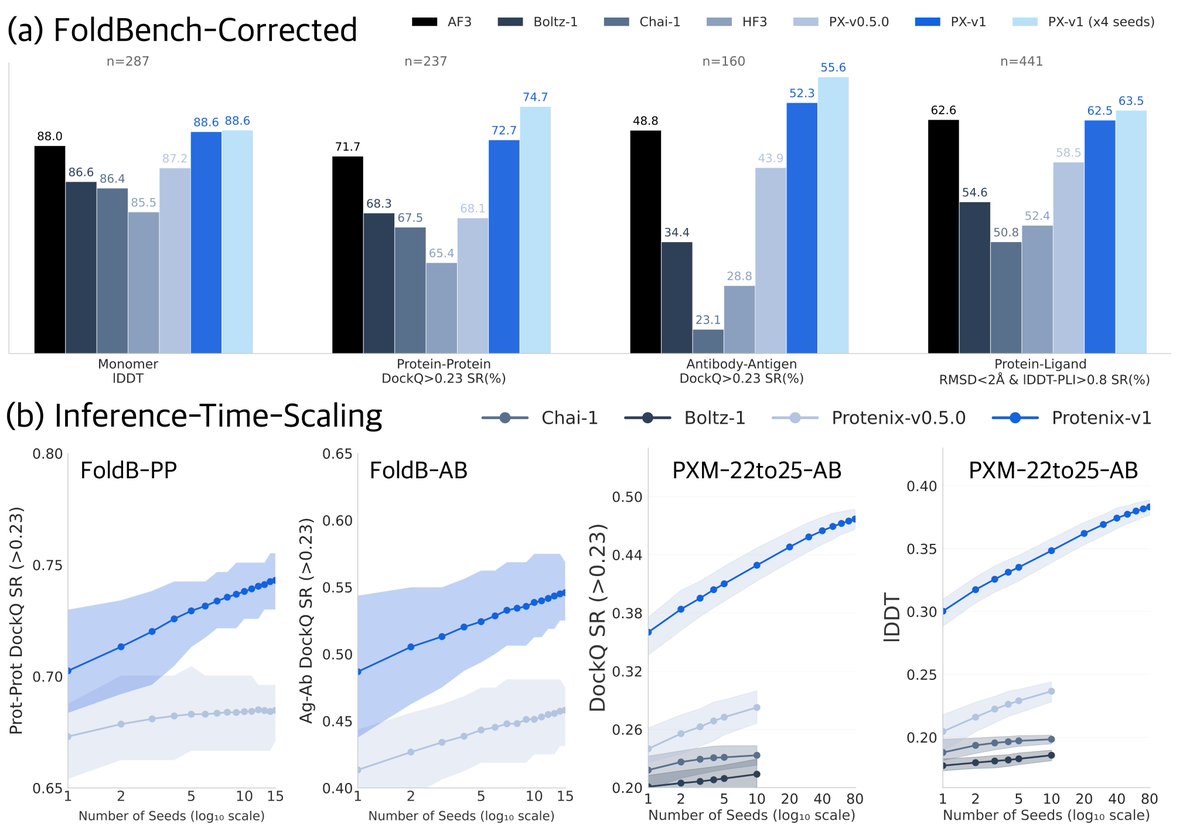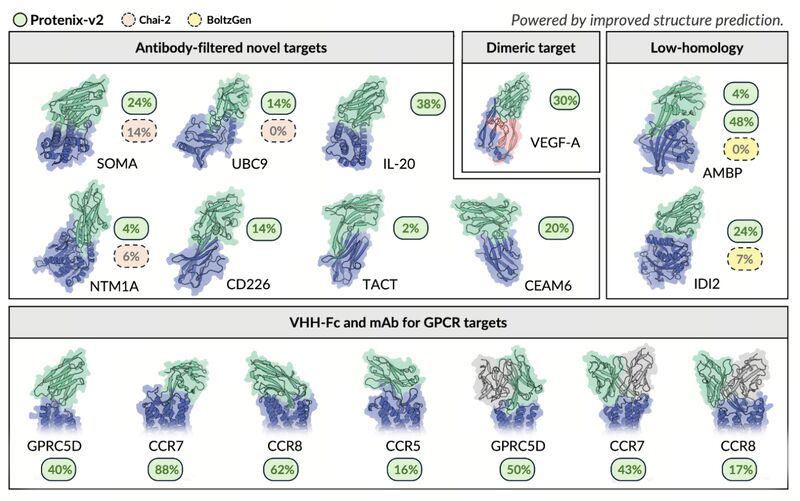

Dheeraj Prakaash retweetledi

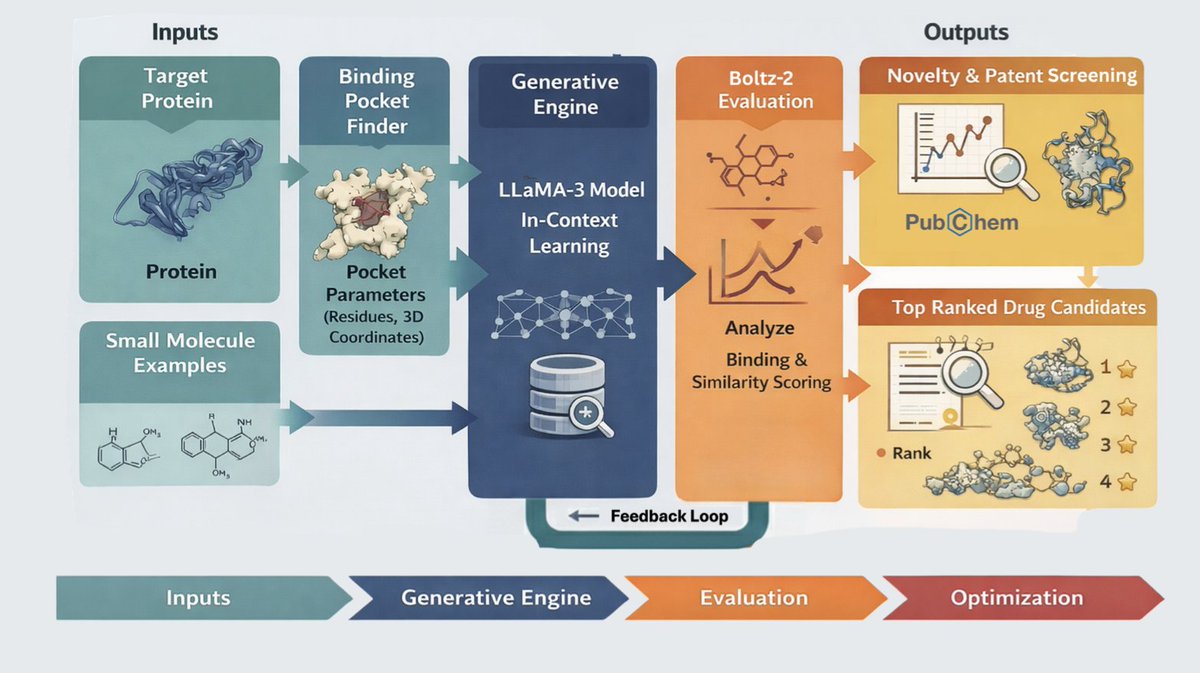
Today, we introduced Gemini For Science, a collection of experimental tools designed to expand the scale and precision of scientific exploration.
Included in Gemini for Science are three (!!!) brand new Google Labs experiments. Meet your new AI research partners: 🧵👇
English