Jake Scott, MD@jakescottMD
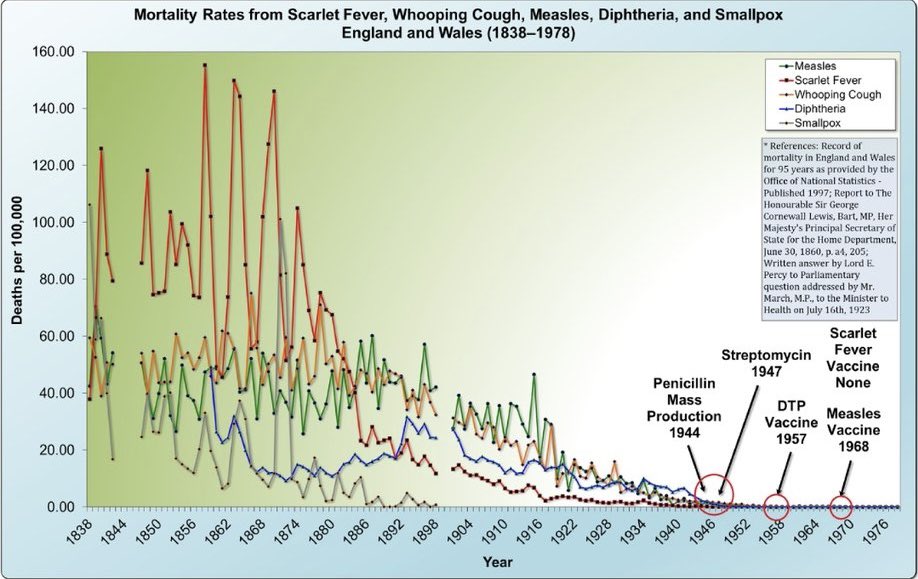
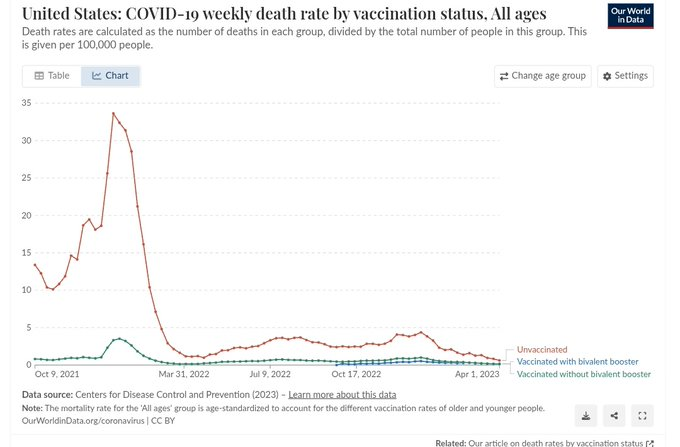
One of the most common problems in the vaccine debate is people citing myocarditis rates that are either inaccurate, outdated, or both. This is a good example.
The 2/10,000 rate (20 per 100,000) claimed here by Dr. Murthy is not supported by any published study I’m aware of. National surveillance in South Korea found the highest dose-specific rate in adolescent males was 5 per 100,000 after dose 2 of BNT162b2 (Ahn 2024, J Korean Med Sci 39:e317). CDC’s Vaccine Safety Datalink, which actively monitors 12-39 year olds, found rates of about 38 per million after dose 2 of the original monovalent vaccine. That was the peak. It’s 3.8 per 100,000, not 20.
But more importantly, that was 2020-2021. The VSD data show that myocarditis incidence dropped with each subsequent formulation and is now back at background levels of about 2 per million for the 2024-2025 doses. A Danish study of over 1 million adults who received the JN.1-adapted vaccine found a myocarditis IRR of 1.12 (95% CI 0.41-3.10), no association (Andersson 2025, JAMA Netw Open 8:e2523557). Two U.S. studies of the XBB.1.5-adapted vaccines also found no signal (Pan 2025, Nat Commun 16:6514; Sun 2025, Vaccine 45:126629).
Citing a historical peak rate, inflating it by 4-5x, and presenting it as if it reflects the current risk is not a serious argument.