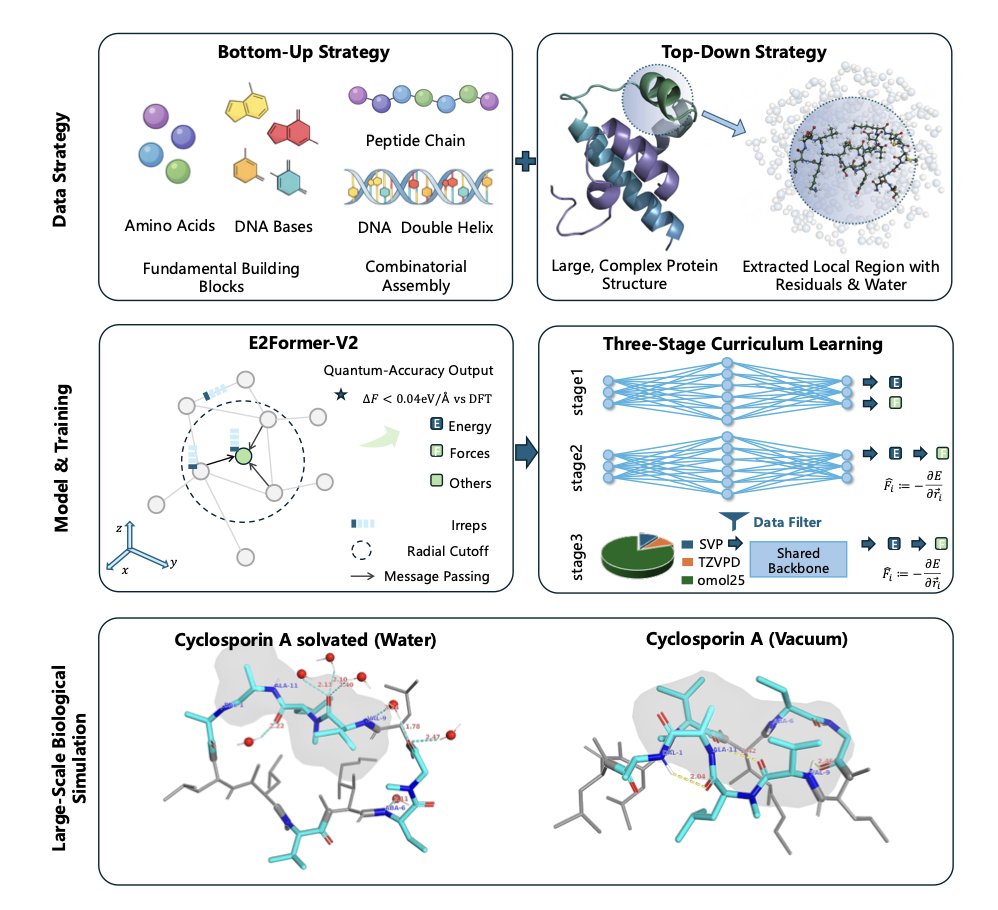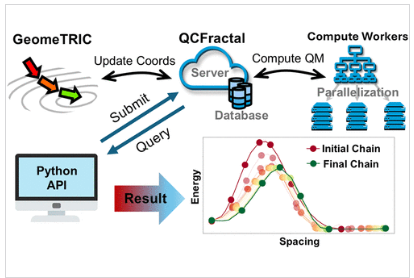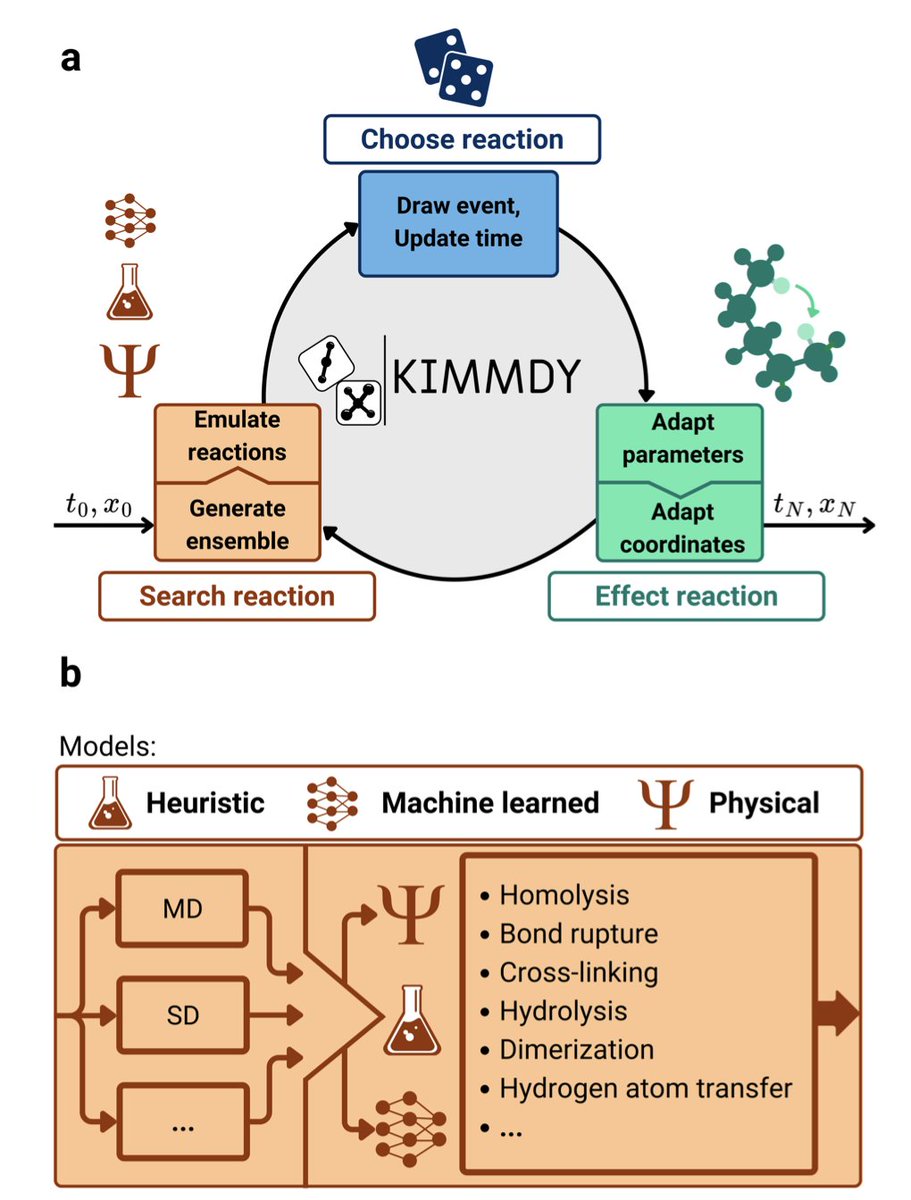

Wayne retweetledi

The TurboQuant paper (ICLR 2026) contains serious issues in how it describes RaBitQ, including incorrect technical claims and misleading theory/experiment comparisons.
We flagged these issues to the authors before submission. They acknowledged them, but chose not to fix them. The paper was later accepted and widely promoted by Google, reaching tens of millions of views.
We’re speaking up now because once a misleading narrative spreads, it becomes much harder to correct. We’ve written a public comment on openreview (openreview.net/forum?id=tO3AS…).
We would greatly appreciate your attention and help in sharing it.
Google Research@GoogleResearch
Introducing TurboQuant: Our new compression algorithm that reduces LLM key-value cache memory by at least 6x and delivers up to 8x speedup, all with zero accuracy loss, redefining AI efficiency. Read the blog to learn how it achieves these results: goo.gle/4bsq2qI
English