Sabitlenmiş Tweet

This is an infrastructure project.
Join us gopetition.com/petitions/a-un…
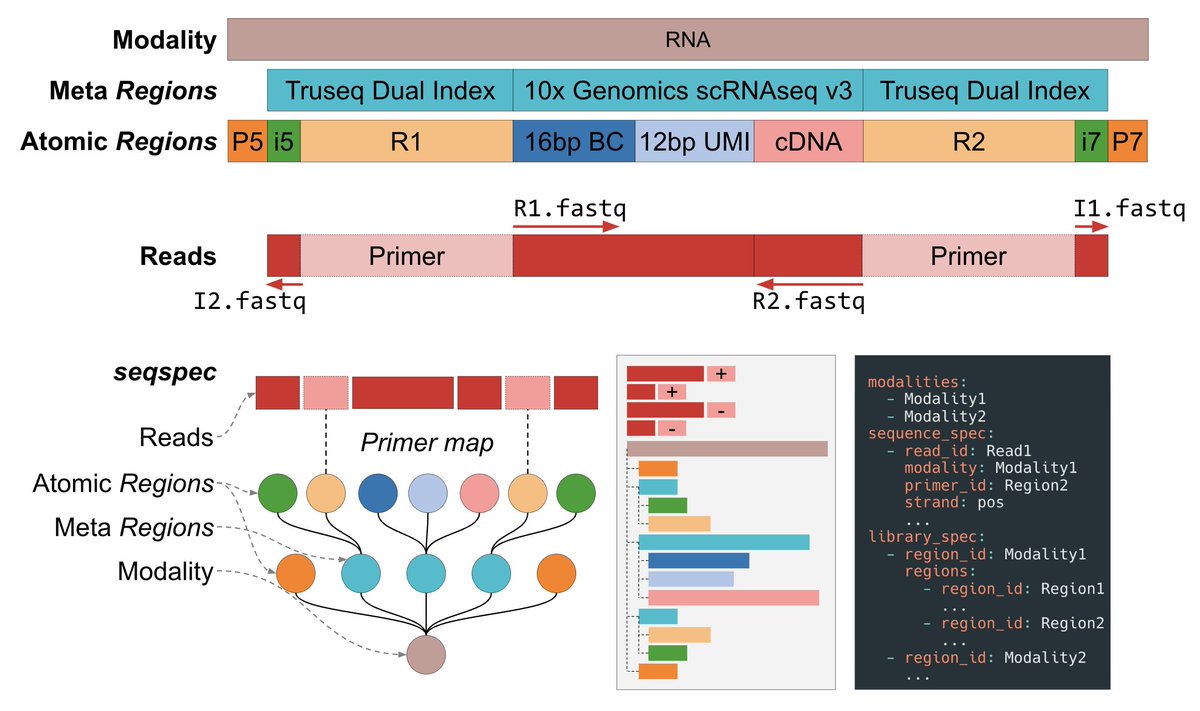
@anshulkundaje @ontowonka 16/16
English
Ben Hitz
1.2K posts
@bonscotthoughts
ENCODE DCC * IGVF DACC (he/him)





@arnavm1 Cell type and state are actually pretty easy to define. Time for a blog post.

In this UMAP, there are arrows linking nothing to nothing (see panel d). Gornisht mit gornisht, as they say. Also drawing curves on top of a UMAP built from x-ray images by repurposing RNA velocity software that already didn't make sense is next level!





I understand that Americans are freaking out and many are thinking about moving out of the States and I would genuinely recommend they do not do that. Fascism is on the rise literally everywhere, esp in Europe. You will not be saved by just moving away.






@vsbuffalo @jgschraiber One of the many terrifying things I learned from your Bioinformatics Data Skills book!