Sabitlenmiş Tweet

Our biggest "side project" so far. Ovo, an open-source ecosystem for de novo protein design, is released today 🧵👇

English
David Prihoda
121 posts

@prihodad
Building tools 🛠️ 🧬 Ovo, BioPhi, DeepBGC. Deep learning, protein design, bioinformatics & random Czech stuff

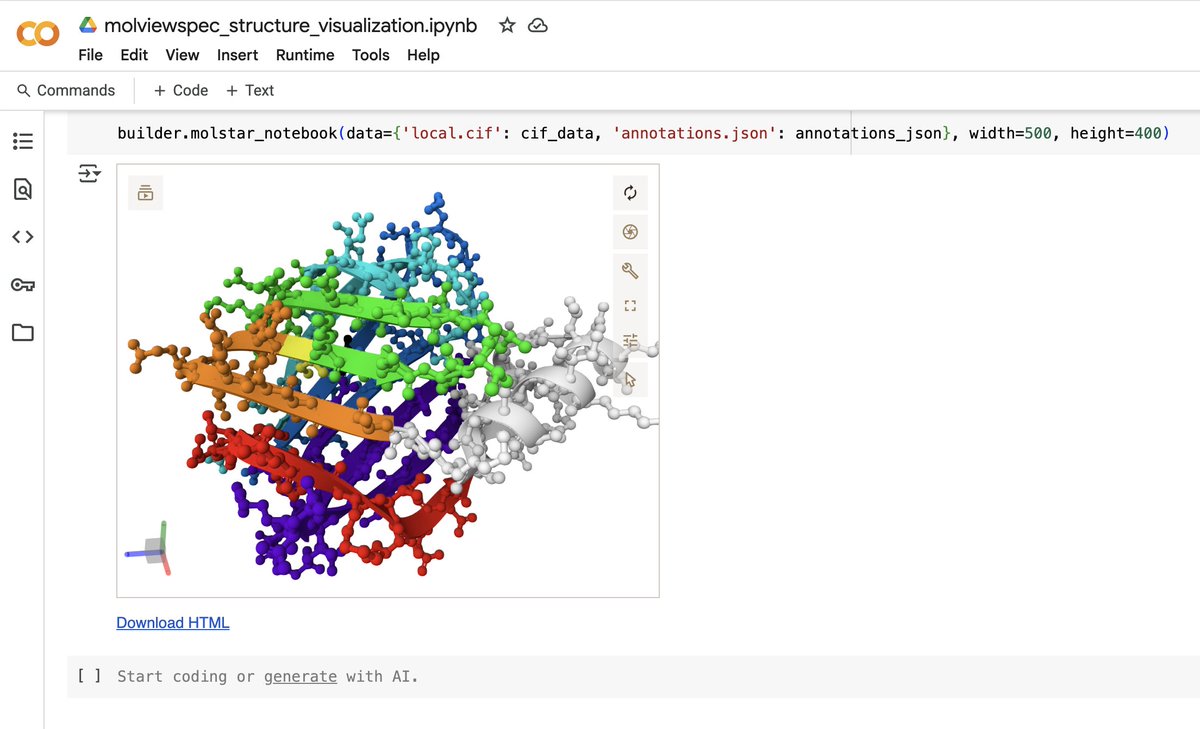
Ovo, an Open-Source Ecosystem for De Novo Protein Design 1. Ovo is a novel open-source platform for de novo protein design, addressing the fragmented landscape of current tools. It integrates models, workflows, data management, and interactive visualization into a scalable ecosystem, making it easier for both experts and non-technical users to design proteins at scale. 2. The platform leverages Nextflow for workflow orchestration, ensuring modularity and scalability across different infrastructures, from local machines to cloud environments. This infrastructure-agnostic design allows for flexible deployment and execution of protein design pipelines. 3. Ovo introduces a novel ProteinQC module that computes comprehensive sequence and structure descriptors, contextualizing designs against reference sets. This feature helps users evaluate the quality and feasibility of their protein designs more effectively. 4. The ecosystem supports scaffold design, binder design, and diversification workflows, with interactive interfaces that simplify the process of choosing appropriate models and submitting jobs. It also includes advanced filtering capabilities to prioritize high-quality candidates for downstream validation. 5. Community-driven development is a core aspect of Ovo, allowing users to add new workflows and plugins. This extensibility ensures that the platform can rapidly adopt and integrate emerging methods, facilitating benchmarking and standardization in the field. 6. Ovo's data management layer ensures efficient organization and retrieval of designs and descriptors, supporting retrospective analysis and linking experimental success rates with computational scores. This robustness is crucial for reproducibility and scalability in industrial settings. 7. The platform's interactive visualization tools enable users to inspect and filter designs based on confidence scores and protein properties, making it easier to identify the most promising candidates for experimental testing. 📜Paper: biorxiv.org/content/10.110… #ProteinDesign #OpenSource #ComputationalBiology #Bioinformatics #Nextflow #DeNovoProteins