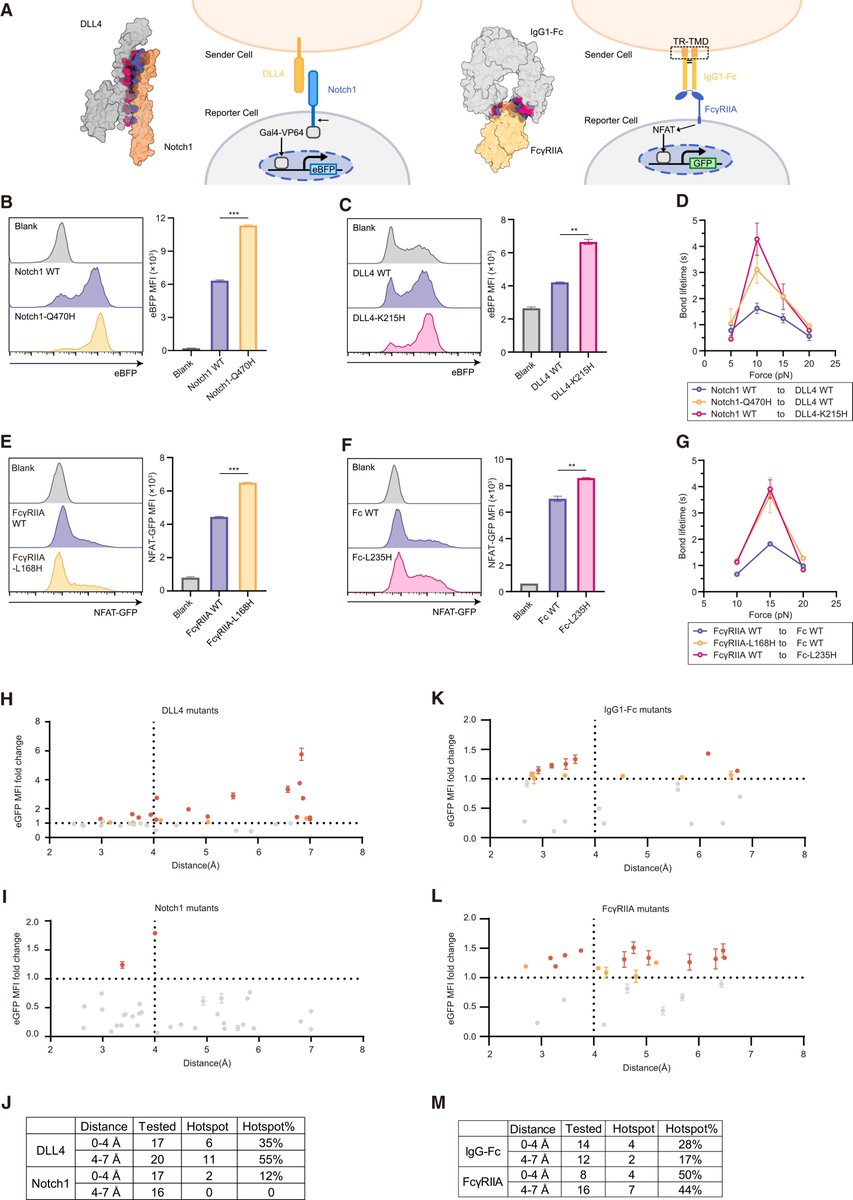
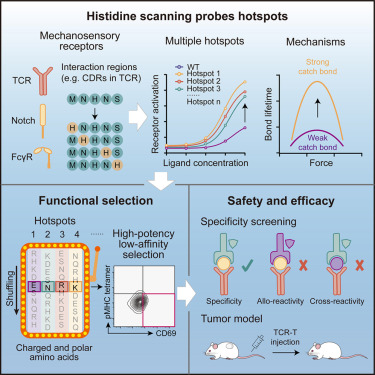
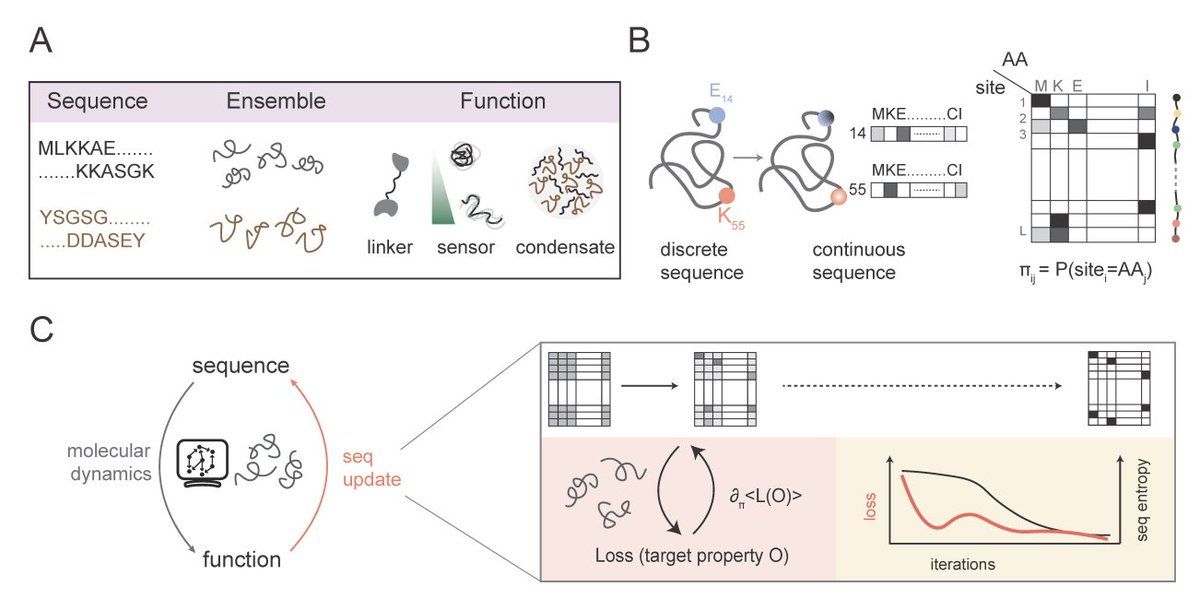
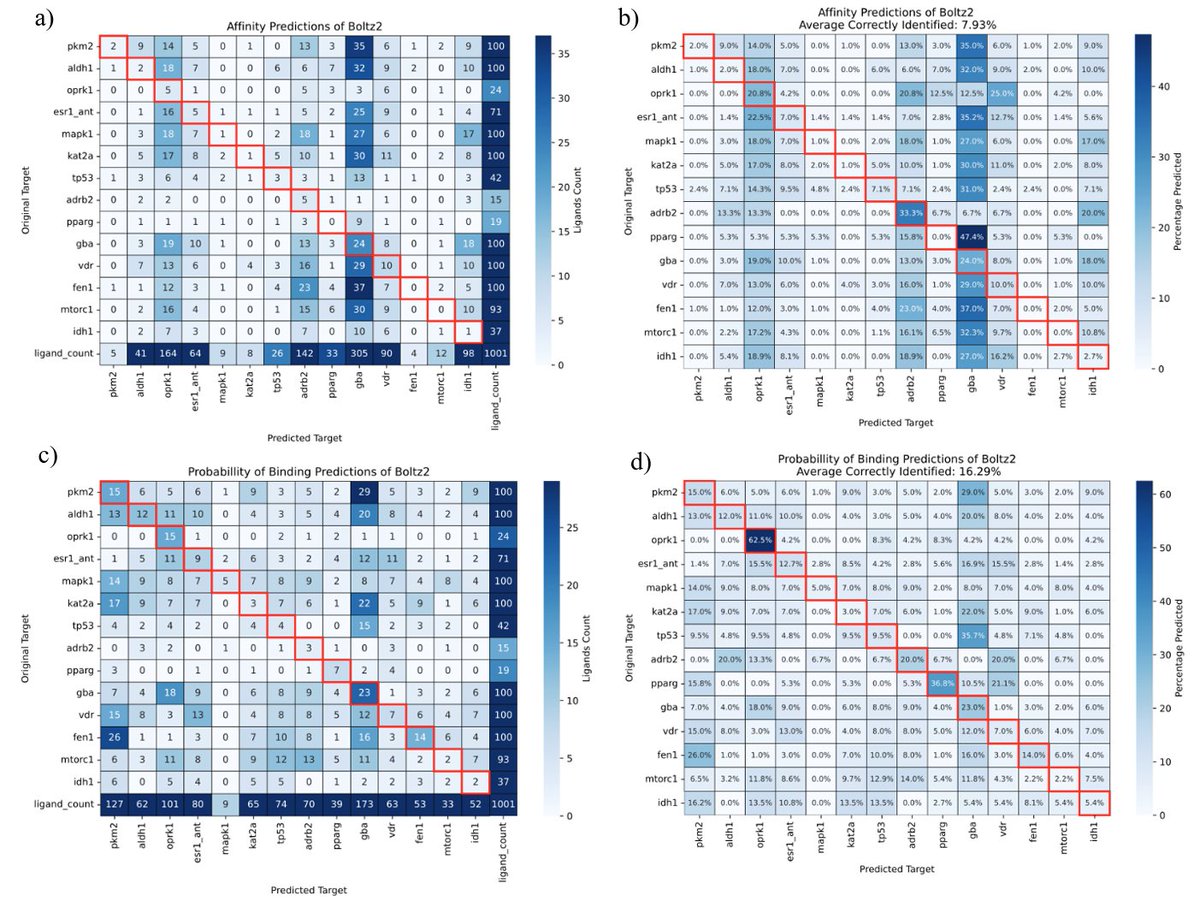
Sabitlenmiş Tweet

Very happy to see our work published!
Synthetic data power!
Thanks for @NatComputSci for their support and to the 5 positive reviewers for feedback!
Thanks to @chevaliersf for writing a very cool News & Views about it too!
Now, Enjoy!
Nature Computational Science@NatComputSci
. @pandaisikit, @probertimmodels, @victorgreiff and colleagues introduce the Absolut! framework, which can generate synthetic 3D-antibody-antigen structures to assist machine learning and dataset construction for antibody design. nature.com/articles/s4358… 👉rdcu.be/c1UcJ
English