Improving AlphaFold2 Performance in Virtual Screens Targeting GPCRs by Enhancing Binding-Site Conformational Sampling
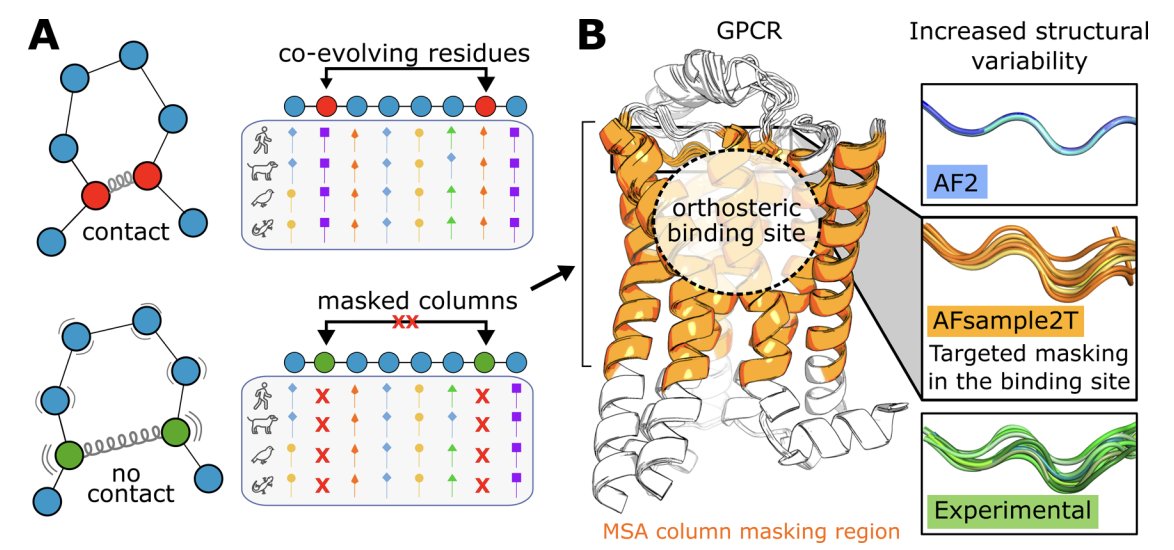
1. The paper introduces AFsample2T, a targeted AlphaFold2 sampling strategy that boosts virtual screening performance for GPCRs by generating diverse binding-site conformations rather than relying on a single “best” structure.
2. Core idea: mask selected MSA columns only in the orthosteric-site region (extracellular-facing TM segments + part of EL2) to weaken local coevolution constraints and encourage alternative pocket geometries, while keeping the rest of the receptor well-constrained.
3. AFsample2T contrasts with global masking (AFsample2), which reduced binding-site accuracy for GPCRs here (AUC 0.38). Targeted masking preserved fold quality while improving pocket modeling, showing that “where to perturb” matters as much as “how much to perturb”.
4. The authors benchmarked 10 class A GPCRs using 61 curated experimental binding-site structures (from an initial pool of 119 PDB structures) and generated 1,000 models per receptor to quantify how often predicted pockets match experimental ones within 1–2 Å side-chain RMSD (symmetry-aware).
5. Moderate targeted masking improved binding-site accuracy: default AF2 AUC 0.54; adding dropout without masking AUC 0.57; targeted masking at 10–30% reached AUC ~0.59–0.61. Too much masking (50%) degraded secondary structure and collapsed performance (AUC 0.43).
6. The best-performing ensemble (AFsample2T) mixes 250 models each from 0%, 10%, 20%, and 30% masking (with dropout), yielding AUC 0.63 and capturing 73.8% of experimental binding sites at 1.5 Å RMSD vs 60.7% for AF2 (a 22% relative gain at that threshold).
7. A key mechanistic improvement is realistic binding-site plasticity. Median binding-site side-chain RMSF increased from 0.15 Å (AF2; overly rigid pockets) to 0.45 Å (AFsample2T), approaching experimental variability (median 0.58 Å). Backbone RMSF similarly moved from 0.10 Å (AF2) to 0.28 Å (AFsample2T), close to experiment (0.30 Å).
8. AFsample2T also mitigates a known docking issue: AF2 often predicts narrow/collapsed pockets. Across receptors, mean pocket volume increased (209 → 218 Å3) and the “top 1% most open” pockets expanded substantially (272 → 389 Å3), closer to experimental pockets (mean 256 Å3). This was especially relevant for μ-opioid receptor, where AF2 pockets were too collapsed.
9. Virtual screening evaluation used DOCK3.8 with ChEMBL actives (52–202 per receptor) and property-matched ZINC20 decoys, totaling extremely large-scale docking (reported as >240 trillion complexes scored). Rigid-receptor docking was used, making pocket microstates critical.
10. Ensemble screening + ligand-guided model selection is the practical win: while median enrichment of AF2-based models remained below experimental structures, the top 1% AFsample2T models improved early enrichment (mean aLogAUC top 1%: 10.8 → 12.9; mean EF1% top 1%: 7.5 → 9.6). In some targets (e.g., TAAR1, μ-opioid receptor), best AFsample2T models approached top experimental-structure performance.
11. The paper provides a workflow for prospective use: generate ≥250 AFsample2T models for the relevant receptor state, dock a ligand/decoy control set to compute enrichment, select ~top 1% models, manually inspect key interactions/poses, then proceed to large-library prospective screening.
12. Modeling receptor state is handled explicitly: inactive models use receptor sequence alone; active models are generated by cofolding receptor with heterotrimeric G protein sequences via AF2-Multimer, capturing hallmark TM6 movements and separating “state sampling” from “pocket microstate sampling”.
💻Code: github.com/wallnerlab/AFs…
📜Paper: doi.org/10.1021/acs.jc…#AlphaFold2#GPCR#VirtualScreening#Docking#StructureBasedDrugDesign#ComputationalChemistry#Bioinformatics#ProteinStructure#DrugDiscovery#MachineLearning
We introduce ConforNets, a mechanism for conformational control in AlphaFold3 models
- SoTA at producing diverse conformations on every multistate benchmark (N=104)
- Novel capability: transfer state from one protein to another
Outperforms BioEmu, ConforMix and AFsample3
🧵1/8
The Iso team has cooked something incredible: our new technical report unveils the latest results from our drug design engine, the IsoDDE, progressing far beyond AlphaFold 3. This breaks new ground compared to AF and other similar methods by a significant degree across all key benchmarks. 1/7
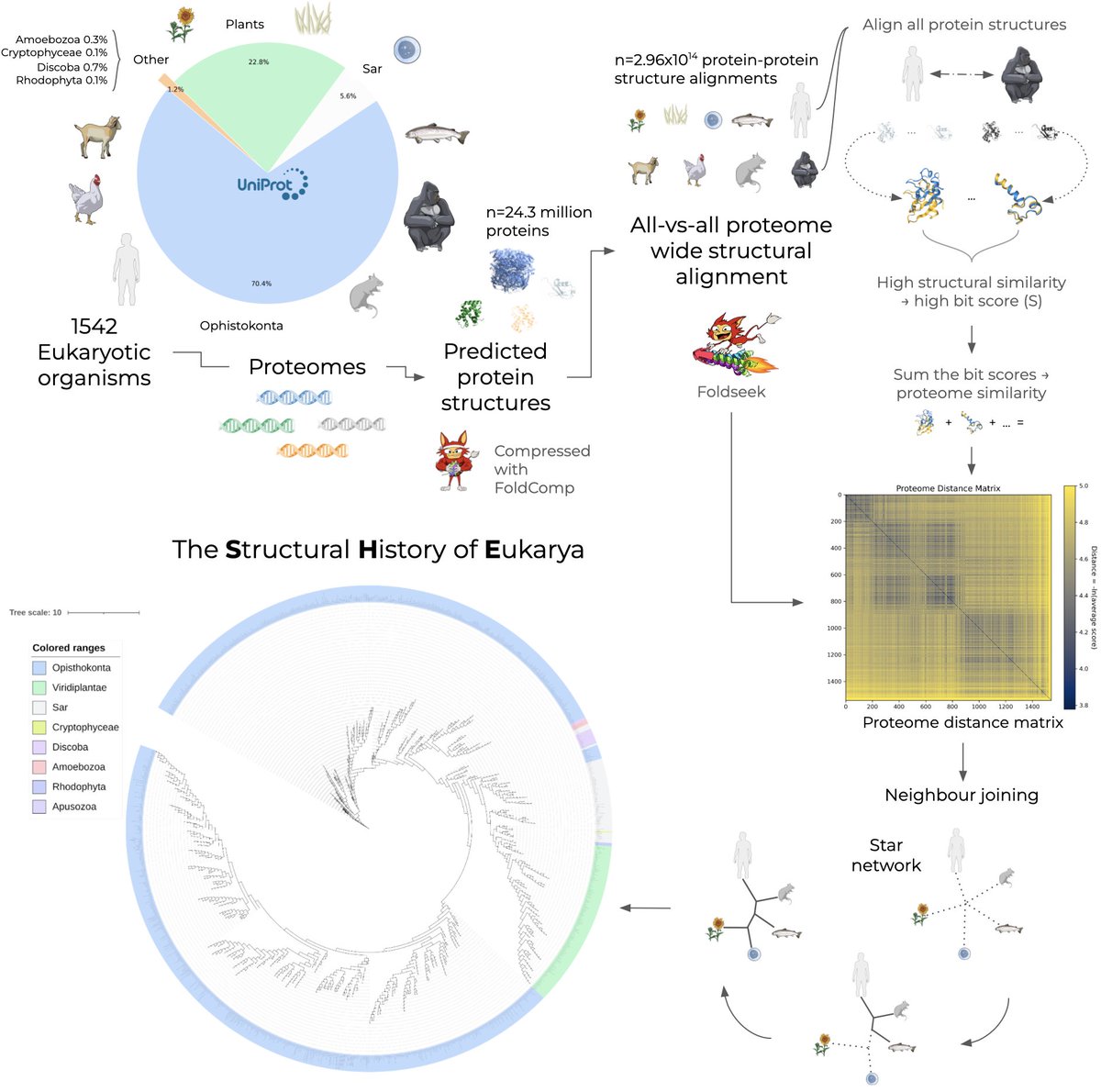
Introducing The Structural History of Eukarya (SHE): The first proteome-scale phylogeny constructed entirely from 3D structure.
We computed 300 trillion alignments across 1,542 species to map the tree of life. 🧵👇 (1/5)
For readers interested in drugs targeting muscarinic acetylcholine receptors, such as Cobenfy, the first drug with a new mechanism for schizophrenia in decades, which is also being investigated in Alzheimer disease, here's a review nature.com/articles/s4157…rdcu.be/e1VOo
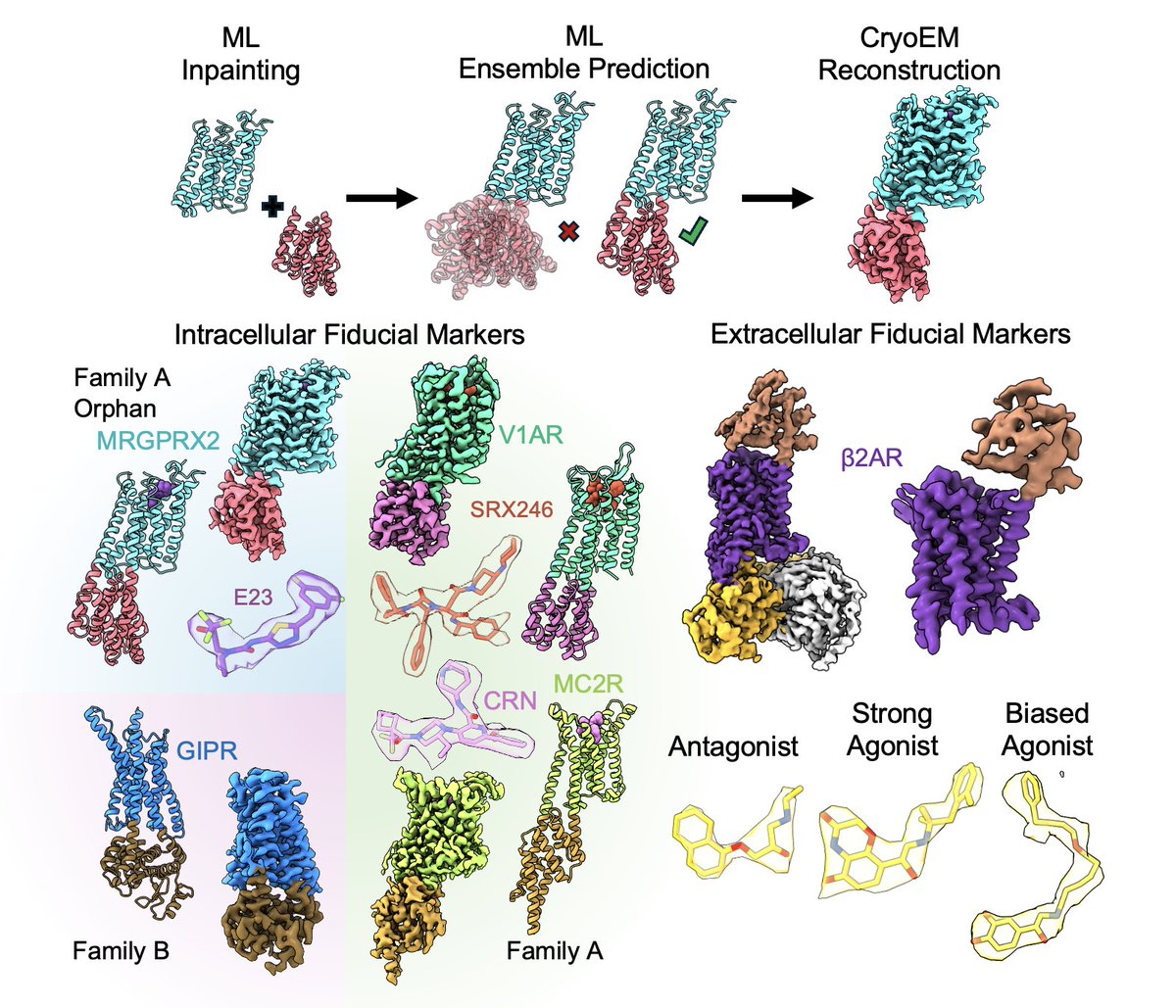
New preprint from the Robertson Lab! We used AF ensemble generation for rigidity filtering with generative design to make cryoEM fiducials, enabling rapid inactive states of four drug targets and a beta2 extracellular fiducial to study GPCR activation!
biorxiv.org/content/10.648…
I thought this was Ai but it’s not.
Yesterday - Northern Lights aka Aurora Borealis over Stonehenge, England.
Simply unbelievable.
Truth truly is stranger than fiction.
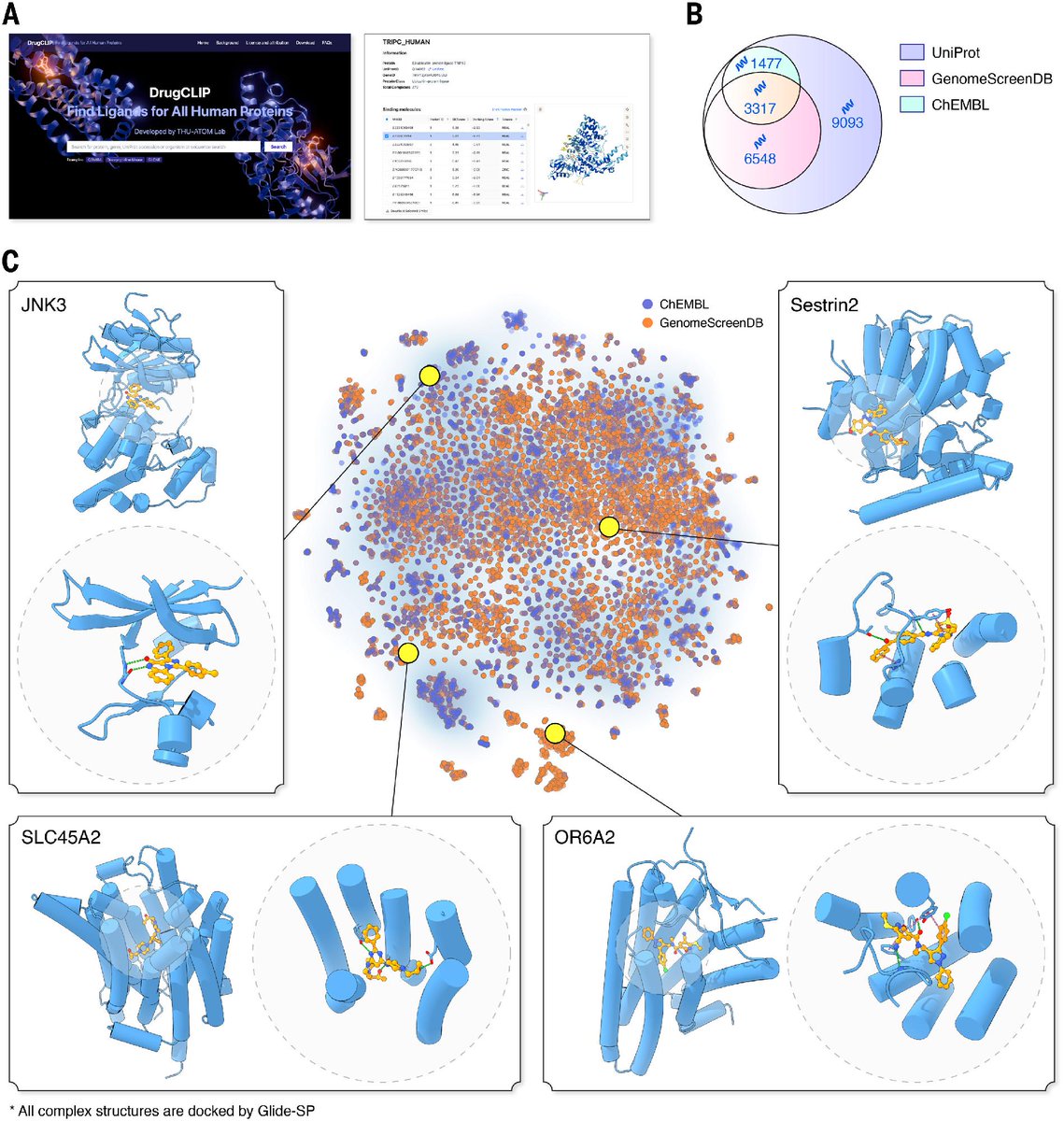
In a new Science study, researchers introduce DrugCLIP, a contrastive learning framework that virtually screens small molecules and protein pockets, analyzing protein-ligand interactions 10 million times faster than most standard molecular docking approaches. scim.ag/49v6Xln
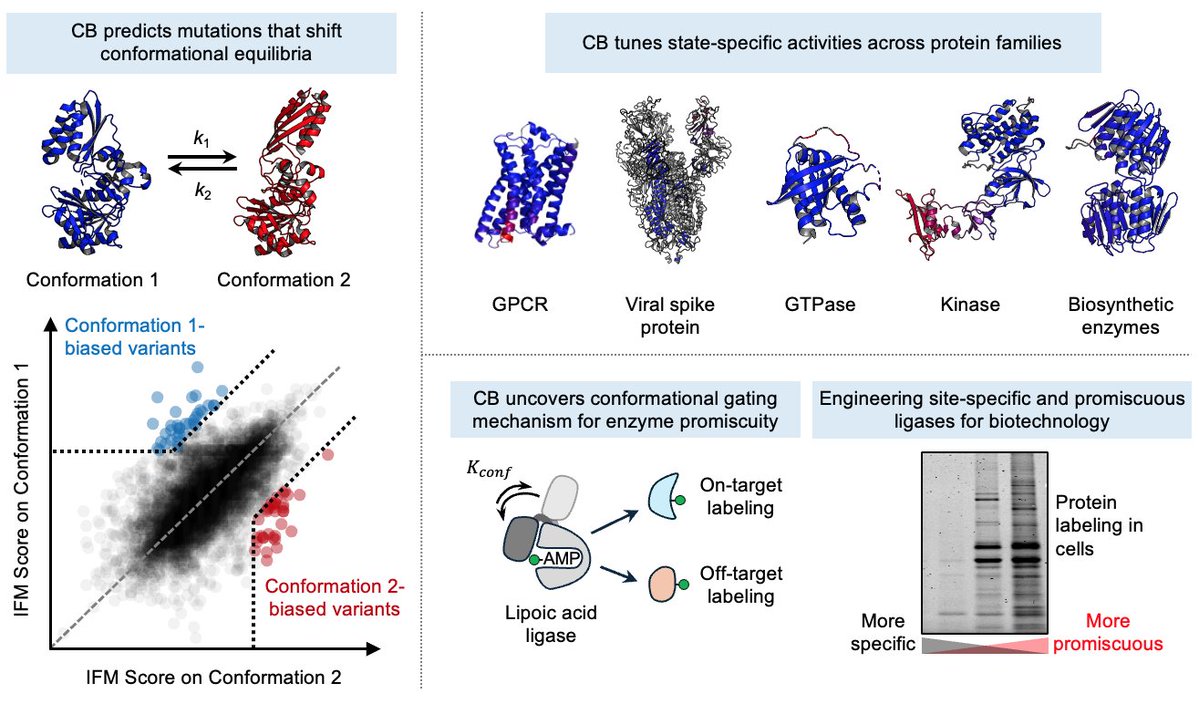
Can we design mutations that predictably bias proteins towards desired conformational states?
Today in @ScienceMagazine, we introduce Conformational Biasing (CB), a simple and scalable computational method that uses contrastive scoring by inverse folding models to identify conformation-biasing mutations.
science.org/doi/10.1126/sc…
Ultra-large virtual screening discovers agonists of the orphan receptor GPR139, a target for schizophrenia. AlphaFold3 fails to model receptor-ligand complexes for understudied targets! New paper: nature.com/articles/s4146…@NatureComms@UU_University@Scilifelab
Big news from Boltz today: we’re launching Boltz Lab, a new platform with new small-molecule + protein design agents, announcing Boltz PBC and a $28M seed round, and sharing a multi-year partnership with Pfizer. More below! 🚀
De novo design of protein competitors for small molecule immunosensing
1. Researchers have developed a novel approach to create protein binders that can compete with small molecules in immunoassays, using the BindCraft pipeline. This method eliminates the need for custom competitor synthesis, which is often time-consuming and analyte-specific.
2. The study leverages deep learning to design binders targeting the antigen-binding sites of antibodies, ensuring competitive binding through predicted steric clashes with small-molecule analytes. This innovative strategy allows for rapid adaptation of immunoassays to different targets.
3. As a proof of concept, the team designed digoxin competitors and experimentally validated them using a bioluminescent assay. They achieved a 70% success rate in identifying functional binders directly from bacterial lysate, demonstrating the efficiency of their approach.
4. Two of the designed binders were integrated into the LUCOS sensor platform, enabling sensitive detection of digoxin with an apparent Kd of 109 nM. This highlights the potential for de novo protein binders to enhance the modularity and sensitivity of existing sensing platforms.
5. The work underscores the power of computational protein design in rapidly generating effective competitors for small-molecule detection. It opens new avenues for developing versatile and adaptable immunoassays for various biomarkers.
📜Paper: biorxiv.org/content/10.648…#ProteinEngineering#Immunoassays#DeepLearning#Bioluminescence#Biosensors
Excited to share @Nature: How does naloxone (Narcan) stop an opioid overdose? We determined the first GDP-bound μ-opioid receptor–G protein (wt) structures and found naloxone traps a novel "latent” state, preventing GDP release and G protein activation. 🧵
nature.com/articles/s4158…