
hinata_ji
120 posts
hinata_ji
@yours_ex_bf
comp bio | building things IISER PUNE'26 trying to write on (will soon) : https://t.co/sp8H5nN5wK
pune Katılım Aralık 2024
376 Takip Edilen8 Takipçiler

🍹Long weekend Project: Since Claude Fable is banned for Science, I thought it might be fun to see if it can be used for something less scientific. 😎
Introducing Age of Epochs! ⚔️ An attempted reproduction of Age of Empires II in Javascript.
ageofepochs.com


English
I am happy to share that our preprint on predicting the structure of chemically modified peptides is out.
I feel so grateful to have been part of the team and to be able to contribute to it.
Preprint link : biorxiv.org/content/10.648…
English
It is finally out
PEPstrMOD2: Next-generation tertiary structure prediction of chemically modified and non-natural peptides biorxiv.org/content/10.648…
English
hinata_ji retweetledi

PEPstrMOD2: Next-generation tertiary structure prediction of chemically modified and non-natural peptides
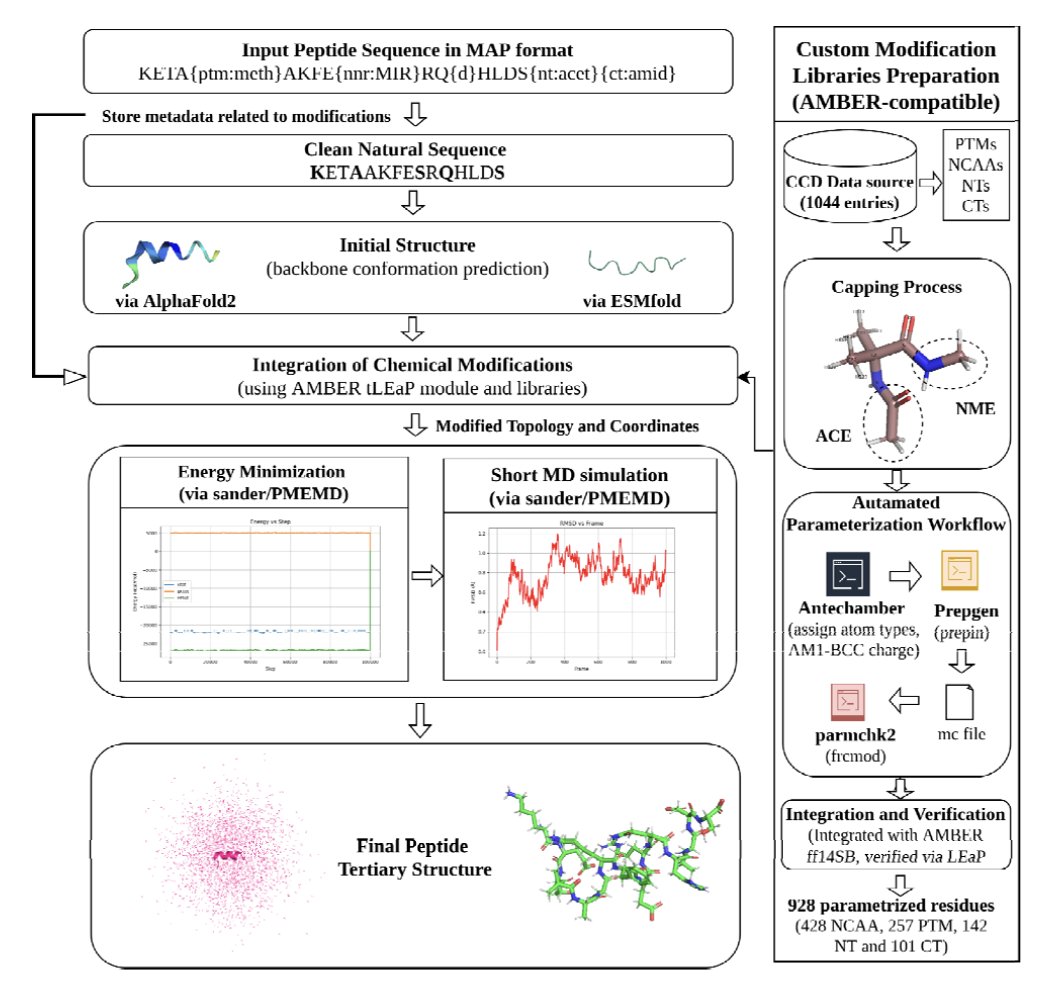
1 PEPstrMOD2 is a structure-prediction framework aimed at chemically modified and non-natural peptides, combining deep-learning backbones (AlphaFold2 or ESMFold) with AMBER-based energy minimization and short MD refinement to restore chemical realism after modifications are introduced.
2 The most practically important upgrade is chemical coverage: an AMBER force-field compatible library parameterized from PDB CCD entries, totaling 928 modifications (257 PTMs, 428 non-canonical amino acids, 243 terminal modifications; including D-residues and cyclic constraints via the input specification).
3 The second major upgrade is scalability: the original PEPstrMOD was limited to 7–25 residues; PEPstrMOD2 removes that restriction by using AF2/ESMFold for initial coordinate generation, enabling modeling of longer therapeutic peptides and small proteins (web UI notes up to 100 residues for submission).
4 The third upgrade is the input/representation layer: MAP inline notation encodes PTMs, NCAAs, terminal caps, D-amino acids, disulfides, and head-to-tail cyclization directly in the sequence, then tLEaP integrates these changes onto the DL-predicted backbone before refinement.
5 Refinement protocol details: 2000-step minimization (1000 steepest descent + 1000 conjugate gradient), then heating/equilibration (50 ps NVT at 300 K + 50 ps NPT at 1 bar), followed by short production MD (user-selectable length; evaluated commonly at 100 ps and 1 ns) in vacuum, explicit water (TIP3P), or a hydrophobic methane box.
6 Cyclic peptide benchmark (AfCyc, n=80; 17 head-to-tail + 63 disulfide-cyclized): with explicit-water refinement, PEPstrMOD2 (AF2-start, average model) reached 2.05 Å mean all-atom RMSD (BB 1.50 Å; CA 1.51 Å), competitive with AfCycDesign (1.82 Å) though behind AF3 (1.13 Å). Water refinement helped more than extending MD from 100 ps to 1 ns.
7 Disulfide geometry check on AfCyc: after refinement, predicted S–S distances clustered around ~1.9–2.1 Å (near the expected ~2.0 Å), and the refinement step corrected problematic cysteine pairing geometries seen in some direct DL outputs.
8 Modified peptide benchmark vs AF3 (ModPep433, n=433; common subset because AF3 could not process all modified residues): PEPstrMOD2 achieved lower mean all-atom RMSD than AF3 (4.49 Å vs 4.67 Å), while also improving over the original PEPstrMOD by a wide margin (and producing much better DL-based starting structures before refinement).
9 Structural quality beyond RMSD on ModPep433: PEPstrMOD2 improved local accuracy (mean LDDT 0.73 vs 0.62 for AF3; far fewer models with LDDT < 0.50), and dramatically improved stereochemistry (MolProbity score 0.94 vs 3.95 for AF3; clashscore 0.34 vs 176.97), indicating the MD/energy-minimization stage is doing important corrective work for modified residues.
10 Structured-peptide subset (ModPep16; ≥60% helix+strand): PEPstrMOD2 roughly doubled accuracy over the original PEPstrMOD (2.50 Å vs 5.84 Å mean all-atom RMSD under 1 ns water refinement). The paper notes several high-RMSD cases are context-dependent (interfaces/ligands/binding-induced structure), emphasizing limits of predicting isolated peptides.
💻Code: github.com/raghavagps/PEP…
📜Paper: biorxiv.org/content/10.648…
#ComputationalBiology #Peptides #ProteinStructure #AlphaFold2 #ESMFold #MolecularDynamics #AMBER #PTM #NoncanonicalAminoAcids #PeptideTherapeutics
English
hinata_ji retweetledi
PEPstrMOD2: Next-generation tertiary structure prediction of chemically modified and non-natural peptides
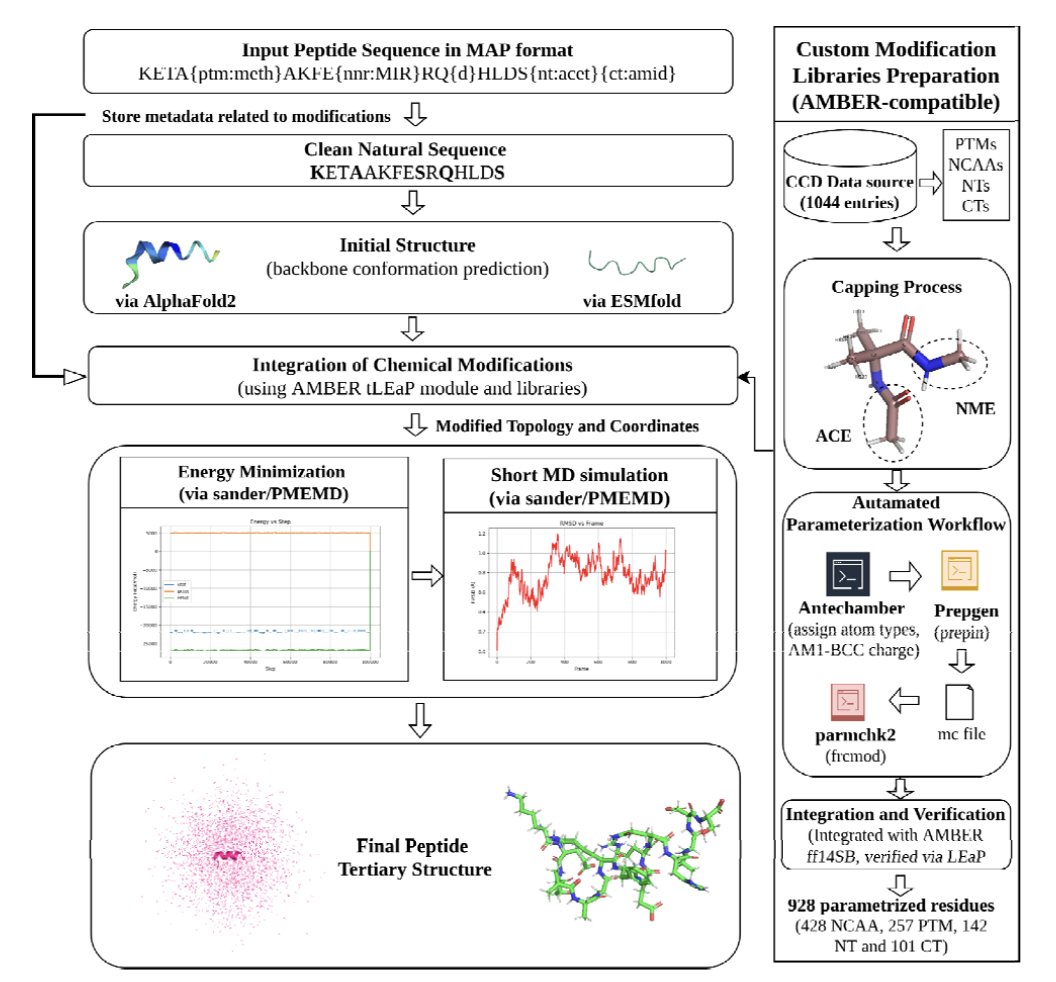
1 PEPstrMOD2 is a structure-prediction framework aimed at chemically modified and non-natural peptides, combining deep-learning backbones (AlphaFold2 or ESMFold) with AMBER-based energy minimization and short MD refinement to restore chemical realism after modifications are introduced.
2 The most practically important upgrade is chemical coverage: an AMBER force-field compatible library parameterized from PDB CCD entries, totaling 928 modifications (257 PTMs, 428 non-canonical amino acids, 243 terminal modifications; including D-residues and cyclic constraints via the input specification).
3 The second major upgrade is scalability: the original PEPstrMOD was limited to 7–25 residues; PEPstrMOD2 removes that restriction by using AF2/ESMFold for initial coordinate generation, enabling modeling of longer therapeutic peptides and small proteins (web UI notes up to 100 residues for submission).
4 The third upgrade is the input/representation layer: MAP inline notation encodes PTMs, NCAAs, terminal caps, D-amino acids, disulfides, and head-to-tail cyclization directly in the sequence, then tLEaP integrates these changes onto the DL-predicted backbone before refinement.
5 Refinement protocol details: 2000-step minimization (1000 steepest descent + 1000 conjugate gradient), then heating/equilibration (50 ps NVT at 300 K + 50 ps NPT at 1 bar), followed by short production MD (user-selectable length; evaluated commonly at 100 ps and 1 ns) in vacuum, explicit water (TIP3P), or a hydrophobic methane box.
6 Cyclic peptide benchmark (AfCyc, n=80; 17 head-to-tail + 63 disulfide-cyclized): with explicit-water refinement, PEPstrMOD2 (AF2-start, average model) reached 2.05 Å mean all-atom RMSD (BB 1.50 Å; CA 1.51 Å), competitive with AfCycDesign (1.82 Å) though behind AF3 (1.13 Å). Water refinement helped more than extending MD from 100 ps to 1 ns.
7 Disulfide geometry check on AfCyc: after refinement, predicted S–S distances clustered around ~1.9–2.1 Å (near the expected ~2.0 Å), and the refinement step corrected problematic cysteine pairing geometries seen in some direct DL outputs.
8 Modified peptide benchmark vs AF3 (ModPep433, n=433; common subset because AF3 could not process all modified residues): PEPstrMOD2 achieved lower mean all-atom RMSD than AF3 (4.49 Å vs 4.67 Å), while also improving over the original PEPstrMOD by a wide margin (and producing much better DL-based starting structures before refinement).
9 Structural quality beyond RMSD on ModPep433: PEPstrMOD2 improved local accuracy (mean LDDT 0.73 vs 0.62 for AF3; far fewer models with LDDT < 0.50), and dramatically improved stereochemistry (MolProbity score 0.94 vs 3.95 for AF3; clashscore 0.34 vs 176.97), indicating the MD/energy-minimization stage is doing important corrective work for modified residues.
10 Structured-peptide subset (ModPep16; ≥60% helix+strand): PEPstrMOD2 roughly doubled accuracy over the original PEPstrMOD (2.50 Å vs 5.84 Å mean all-atom RMSD under 1 ns water refinement). The paper notes several high-RMSD cases are context-dependent (interfaces/ligands/binding-induced structure), emphasizing limits of predicting isolated peptides.
💻Code: github.com/raghavagps/PEP…
📜Paper: biorxiv.org/content/10.648…
#ComputationalBiology #Peptides #ProteinStructure #AlphaFold2 #ESMFold #MolecularDynamics #AMBER #PTM #NoncanonicalAminoAcids #PeptideTherapeutics
English


@eigenron @prfctsnst 1. Today -> death
2. Today -> experiences -> death
So if it you think the experience is worth it then not dying make sense.
English
@eigenron @prfctsnst Let's assume death as state 'n' which is independentof time, and one will reach that state either today or after sometime anyways, and if we die to day or tomorrow doesn't matter since that state 'n' will remain like that. So we are left woth two ways :
English
hinata_ji retweetledi

When I was a young PhD student I realized the difference between chemistry and biology training at the time as that the former was completely devoid of the concept of controls and variability. Chemists writing biology papers would do n=1 experiments, without confidence intervals or negative controls. (May be different now.)
History repeats now with CS bros expecting they can feed some omics data from one cultured cell line into their model and they will learn how all cells work.
English
@michellearning @DARPA @NVIDIAHealth wet lab data being interpret using ai tools is big thing, I don't think these tools are good enough own their own, they still need a researcher in loop, ai agent can be a junior partner not researcher itself, btw congratulations
English

The future of bio is powered by faster data
Introducing the Medra AI Experimentalist: an agent that turns goals into experimental designs, learns from every result, and develops the next assay
Excited to collaborate with @DARPA and @NVIDIAHealth on the future of science
English

A bit of news: After nearly 9 years, I have decided to leave Google DeepMind and join Anthropic (after taking some time to recharge). I am incredibly grateful for my time at GDM. @demishassabis took a real chance letting me lead the AlphaFold team just six months after finishing my PhD, and the entire GDM team taught me so much about how to do great science. GDM is a special place, and I’ll still be excited to hear about what amazing things they discover next.
English

Was studying the internals AF3 and all i can say, alphafold 3 is much simpler than af2.

English