
Both comments are very relevant
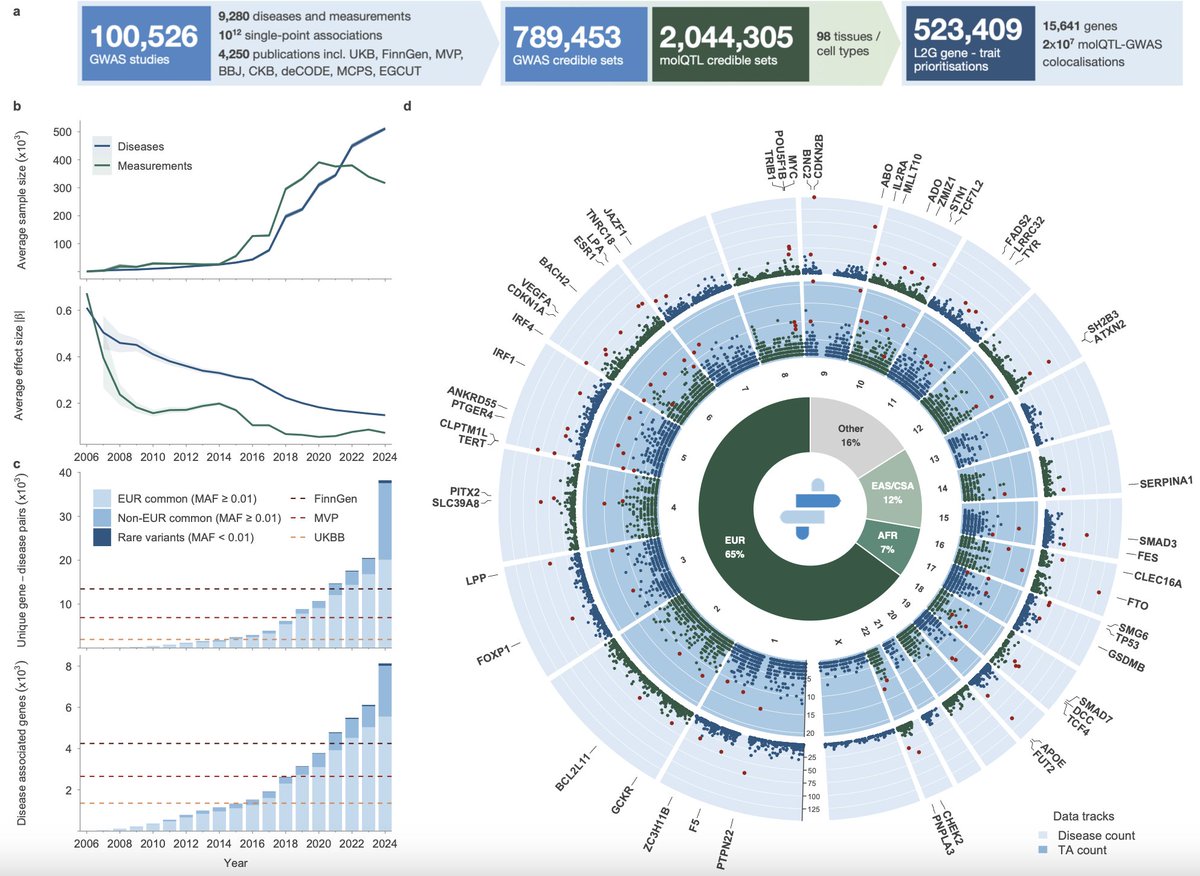
Regarding variant-to-gene mapping. Main conclusions of the paper remain more robust if we focus on PAVs (protein-altering variants). The authors separate PAVs as a distinct category in many analyses, and their mapping to genes is much less ambiguous than for other GWAS signals.
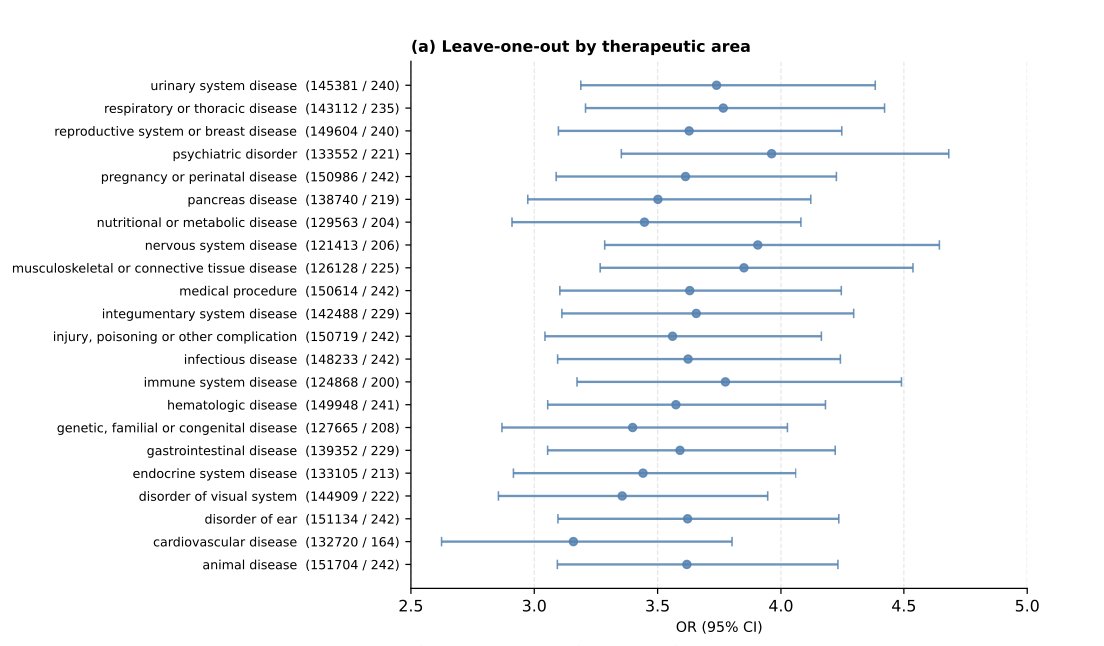
On the second point. Yes, authors did sensitivity analysis by excluding individual therapeutic areas. Overall, the results remained stable, suggesting that the signal was not driven by a single TA. Still it would be very interesting to look at the effects within each therapeutic area separately.
They also used a mixed-effects logistic regression to account for heterogeneity across TAs. The effect size decreased, but not dramatically — from OR = 3.62 to OR = 3.14.
English