
English
Rasmus H
1.8K posts

@RazBioTech
Founder @ https://t.co/rvCWR16DUM | 🧬 #biotech retail investor | 👀 $OCUL maxi | 🌎 Global citizen | 👋 always happy to connect







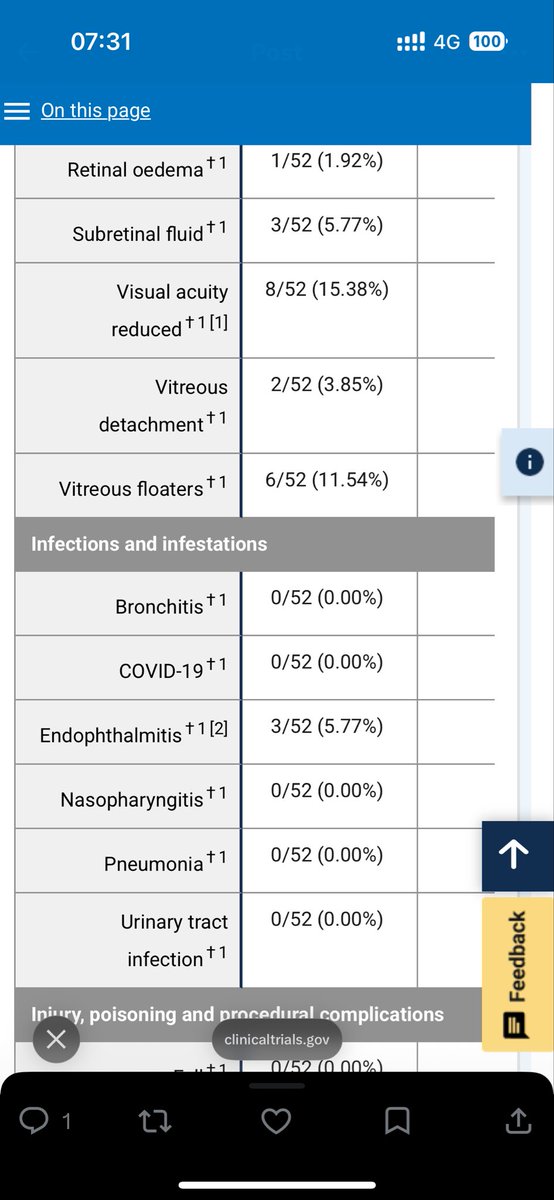
$ocul @zipjet Looking at the FDA NDA process below, the one area that IMO, will require additional data is the Dosing/Safety (with respect to redosing) label negotiation at the end of the process. I believe SOL-1 easily satisfies all of the other requirements. SOL-1 does a good job with efficacy and general safety, but doesn't answer the question about redosing and safety associated with redosing, or efficacy for that matter if you want to be a stickler. SOL-R data could be submitted in February, falling within the 2-7 month window for additional information. So, IMO... the FDA will accept the NDA and will require SOL-R data for final label dosing and safety determination. NDA PROCESS....... 1. Filing Review (First ~60 days) July1 - Sept. 1 After submission, the FDA checks whether the NDA is complete enough to review. Key steps: -Administrative completeness check -Verification that all required clinical, safety, manufacturing, and labeling data are included. -Decision to “file” or refuse to file If accepted, the FDA assigns a PDUFA target action date under the Prescription Drug User Fee Act. That date becomes the official review deadline. 2. Primary Scientific Review (Months 2–7) Sept. 1 - March 1 Multiple FDA teams review different parts of the application simultaneously: Clinical Review -Evaluates whether the drug actually works for the proposed indication. -Reviews clinical trial design, endpoints, and statistical validity. Statistical Review -Re-analyzes trial data to confirm results. Safety Review -Looks at adverse events and long-term safety signals. Pharmacology/Toxicology -Reviews animal and mechanistic data. CMC (Chemistry, Manufacturing, and Controls) -Ensures the drug can be manufactured consistently and safely. 3. FDA Questions to the Company (Throughout Review) During the review, the FDA sends information requests (IRs) or discipline review letters. The company must: -Provide additional data -Clarify analyses -Sometimes submit new manufacturing information or stability data These exchanges can happen multiple times. 4. Facility Inspections (Typically Months 6–9) Dec. 1 - March 30. The FDA inspects: -Drug manufacturing plants -Clinical trial sites -Testing laboratories These inspections verify: -Good Manufacturing Practices (GMP) -Data integrity -Quality systems 5. Advisory Committee Meeting (If Needed) For some drugs, the FDA convenes an external expert panel, such as an advisory committee. Experts discuss: Clinical benefits Safety concerns Risk–benefit balance They then vote on whether the drug should be approved. The FDA is not required to follow the vote but often does. 6. Labeling Negotiation (Late Review Phase) The FDA and the company negotiate the drug label, including: -Approved indication -Dosing -Safety warnings -Contraindications -Risk mitigation programs (if needed) The label determines exactly how the drug can be marketed. 7. Final FDA Decision (PDUFA Date) Apr/May






