The Sharing Scientist retuiteado

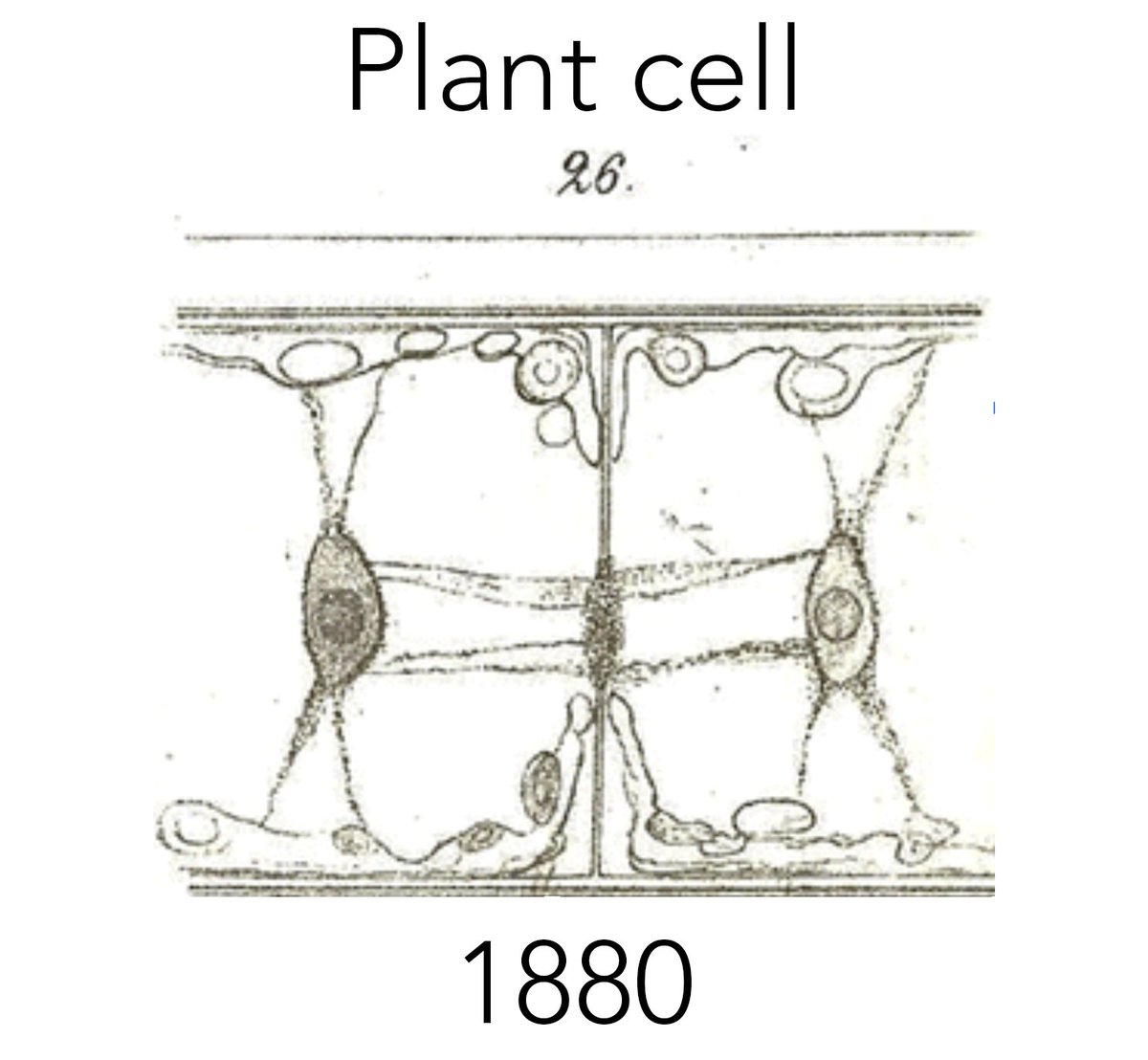
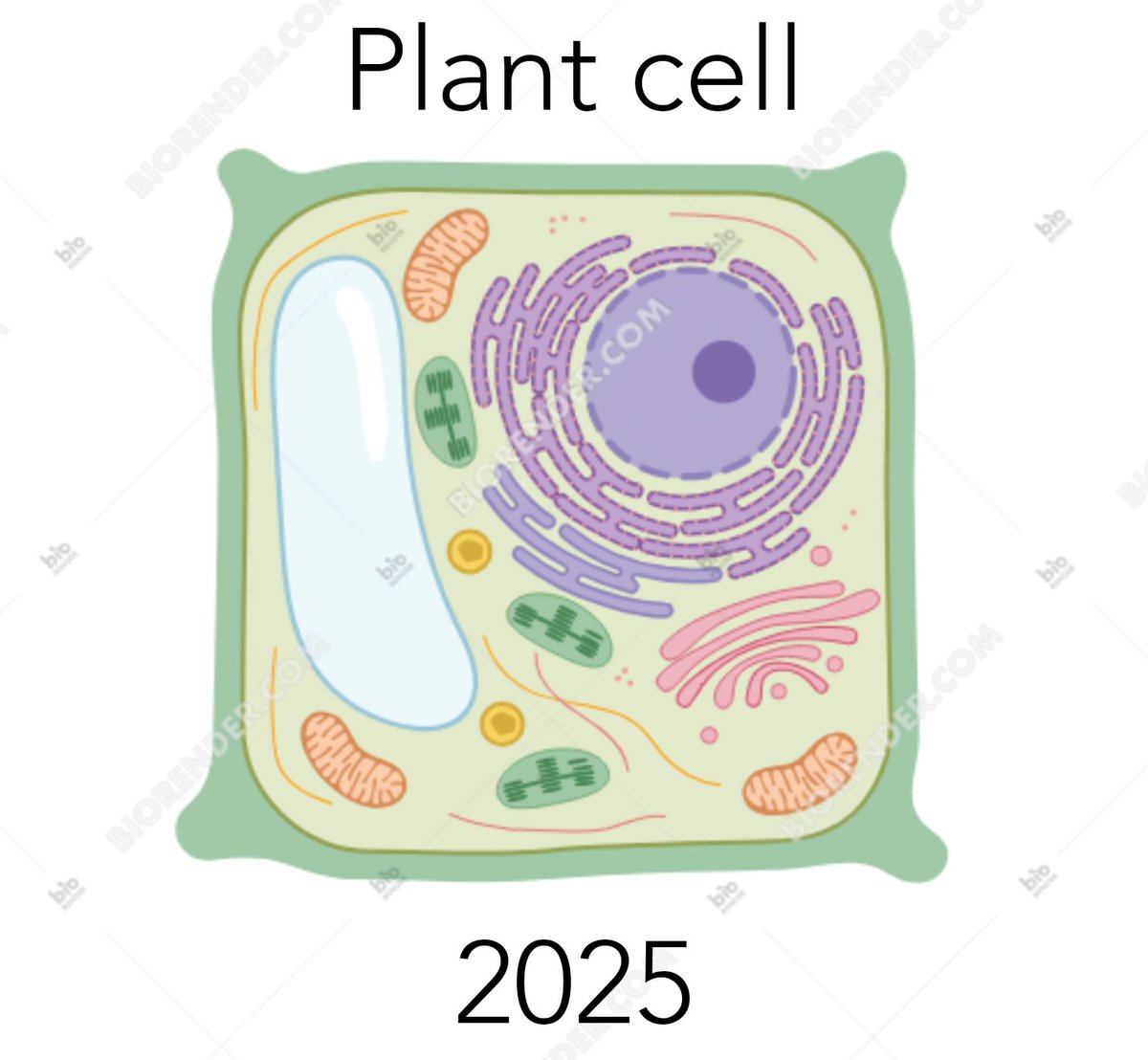
A periodic reminder to leave behind simplistic & naive notions.

English
The Sharing Scientist
14.9K posts


@ScienceShared
Bringing you the most interesting & important science news that you need, summarised & sourced directly to your feed. Also will call out stuff others don't.

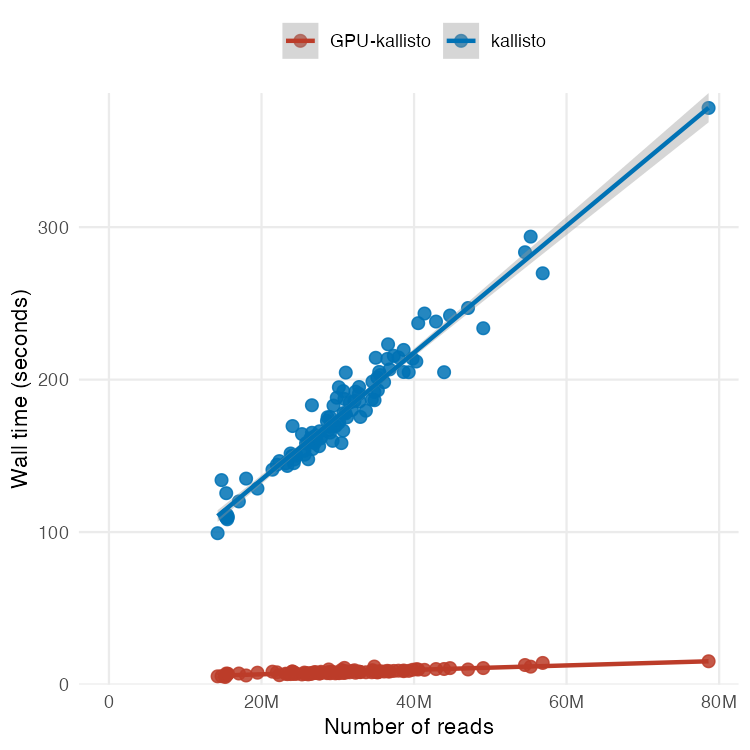

🚨: Scientists mapped 1 mm³ of a human brain ─ less than a grain of rice ─ and a microscopic cosmos appeared.




@lifebiomedguru @LocasaleLab And a healthy dollop of actual analysis on the historical foundation of the field one is trained in, along with critical philosophy and classic logic. Knowing where you information comes from, along with the foundational assumptions, will lead to far fewer dead ends.






Looking for one more high here to test or come close to ATH before pulling some and going sideways for a few days. NFA/NTA








"Only ~0.02%-3.1% of [a cell's] genome" is being transcribed at any given moment. Other interesting takeaways from this new paper: > If you pool together a bunch of cells of the SAME type (like primary immune cells from a mouse's spleen), and you measure the transcription for each of them, you'll find that ~67% of the genome is active collectively. But at a SINGLE cell level, only like 0.04% of the genome is active. There is huge heterogeneity between cells, even of the same type. This heterogeneity disappears when we do bulk RNA-seq and measure cells together. > About 31% of a cell's transcription comes from known protein-coding genes. The rest of transcription happens in regions that don't make proteins. In other words, more "non-coding" DNA is transcribed than "coding" DNA. > There is a surprising disconnect between RNA production & decay at the single-cell level. If you look at thousands of cells together, the rate of RNA production (how fast genes are transcribed) usually matches the rate of RNA decay (how fast old transcripts are degraded). This makes sense, because cells presumably would want to keep a fairly steady balance of RNA levels. But when scFLUENT-seq was used to look at individual cells, this "rule" broke down! For a given mRNA, some cells were making a lot of new copies even if old copies weren’t being degraded much, while other cells had the opposite. So transcription and decay don't seem to be tightly matched within a single cell at a given time after all. The balance between production + decay is only true in bulk.