Sabitlenmiş Tweet

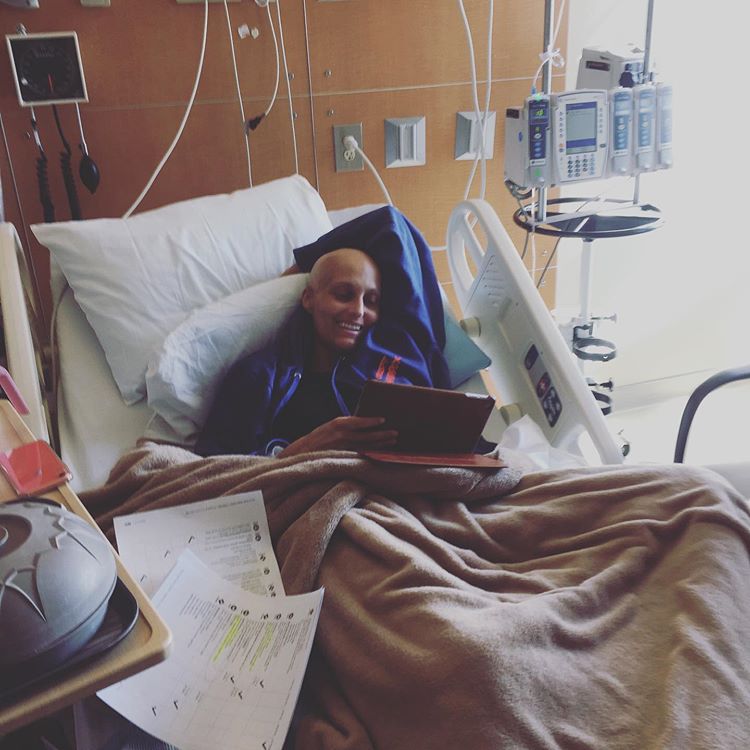
My wife @ElanaMD is an unstoppable force. A month in a small hospital room—chemo, radiation, and a bone marrow transplant— and she still manages to smile through it all.
English
Daniel Tawfik
3.7K posts

@dantawfik
Founder and developer https://t.co/G5rNcIMx4t. I write @ https://t.co/L0KR2JkKbu. Molecular biologist turned dev. product of big government @ucla



30% of cognitively healthy people have amyloid plaques. Yet, they show no signs of Alzheimer's. This has forced a fundamental question: what if amyloid is a symptom, not the cause? Dr. Francisco Gonzalez-Lima's research suggests the real driver may be a metabolic collapse that begins years—even decades—before plaques form. In postmortem studies, the posterior cingulate cortex showed up to 39% reductions in cytochrome c oxidase activity compared to controls. This wasn't secondary to amyloid pathology. It preceded it. Cytochrome c oxidase (CO) is the terminal enzyme in mitochondrial respiration. It transfers electrons to oxygen, generating the proton gradient that powers ATP synthesis. When CO activity drops by 30–40%, neurons can't sustain baseline energy demands. Synaptic transmission weakens, cellular repair stalls, and oxidative stress accumulates. The posterior cingulate cortex is particularly vulnerable—it's metabolically expensive, densely connected, and shows hypometabolism on PET scans years before cognitive symptoms emerge. Dr. Gonzalez-Lima frames Alzheimer's as a vascular-hypometabolic disorder. Reduced cerebral blood flow limits oxygen and glucose delivery. Mitochondria fail. Neurons shift to less efficient metabolic pathways, triggering a cascade that eventually includes amyloid deposition—not as the initiating event, but as a downstream consequence of energy failure. This repositions the therapeutic target. Instead of clearing plaques after neurodegeneration has advanced, the goal becomes restoring mitochondrial function and cerebral perfusion before irreversible damage occurs. Two approaches show mechanistic alignment: Methylene blue acts as an electron shuttle, donating electrons directly to cytochrome c and bypassing damaged segments of the electron transport chain. At 0.5–4 mg/kg, it increased oxygen consumption by 37–70% and ATP production by ~30%. Ketones bypass impaired glucose metabolism entirely, supplying acetyl-CoA directly to mitochondria. Research by Dr. Stephen Cunnane shows ketones can provide up to 60% of the brain's energy needs, even when glucose utilization is compromised. Both target the energy deficit without requiring amyloid clearance. The metabolic overlap is striking: 8 out of 10 Alzheimer's patients also have type 2 diabetes or abnormal glucose levels. Meta-analyses link diabetes to a 1.25–1.91-fold increased dementia risk. This suggests shared pathophysiology—insulin resistance, vascular insufficiency, mitochondrial dysfunction—long before cognitive symptoms become clinically apparent. If the vascular-hypometabolic cascade begins decades early, interventions targeting cerebral blood flow and mitochondrial resilience may alter disease trajectory in ways that amyloid-focused therapies have not. This week's Research Review examines the evidence behind methylene blue, ketones, and the hypothesis that Alzheimer's may be fundamentally a disorder of energy metabolism. gethealthspan.com/research/artic…



After age 60, resting metabolic rate declines by approximately 0.7% per year—even when body composition remains stable. That's roughly 10 calories per year for most adults. Over a decade, it compounds to 100 fewer calories burned daily. The decline accelerates with age and can't be explained by muscle loss alone. Pontzer's work on the doubly labeled water method—measuring total daily energy expenditure in free-living populations—revealed that metabolic rate stays remarkably stable from age 20 to 60 when adjusted for body size and composition. The decline begins after 60. And it's consistent across populations, regardless of activity level or geographic location. This suggests the mechanism isn't behavioral. It's biological. Three factors drive the post-60 decline: organ mass reduction, mitochondrial inefficiency, and chronic inflammation. Metabolically active organs shrink with age. The liver, brain, heart, and kidneys collectively account for roughly 60% of resting energy expenditure despite representing less than 6% of body weight. When these organs lose mass or reduce metabolic activity, basal caloric burn drops significantly. The liver is particularly important. It's responsible for gluconeogenesis, protein synthesis, and lipid metabolism—all energy-intensive processes. A 10% reduction in liver mass can reduce resting metabolic rate by 2%, independent of muscle loss. Mitochondrial function also declines independently of mitochondrial quantity. The electron transport chain becomes less coupled—meaning more oxygen is consumed to produce the same amount of ATP. Reactive oxygen species increase. Membrane potential destabilizes. ATP production efficiency drops. This isn't about having fewer mitochondria. It's about the existing mitochondria working less effectively. Chronic low-grade inflammation compounds both effects. Elevated IL-6, TNF-α, and other pro-inflammatory cytokines create a cellular environment that dampens energy expenditure. Even when muscle mass and organ function are preserved, inflammation reduces metabolic rate at the cellular level. The practical implication: maintaining muscle mass after 60 is necessary but not sufficient to prevent metabolic decline. Resistance training preserves muscle. But it doesn't address organ shrinkage, mitochondrial inefficiency, or inflammation. Those require aerobic exercise for mitochondrial function, dietary interventions for inflammation control, and potentially metabolic optimization strategies targeting insulin sensitivity and nutrient sensing pathways. The 0.7% annual decline is modest in any single year. But over two decades, it compounds to a 14% reduction in resting metabolic rate—roughly 200–250 fewer calories burned daily for an average adult. That's the metabolic equivalent of skipping a meal every two days without changing intake. Our latest Research Review examines the post-60 metabolic trajectory and why multi-layered interventions targeting muscle, mitochondria, and inflammation may slow—but not fully prevent—the decline. gethealthspan.com/research/artic…

Methylene blue doesn't work like most drugs. It doesn't bind to a receptor or block an enzyme. It cycles electrons—donating them to cytochrome c when mitochondria are under stress, bypassing damaged segments of the electron transport chain and sustaining ATP synthesis. In Alzheimer's disease, cytochrome oxidase activity drops by up to 39% in the posterior cingulate cortex—often decades before plaques appear. This week's Research Review examines how methylene blue targets the energy bottleneck that may precede neurodegeneration. gethealthspan.com/research/artic…