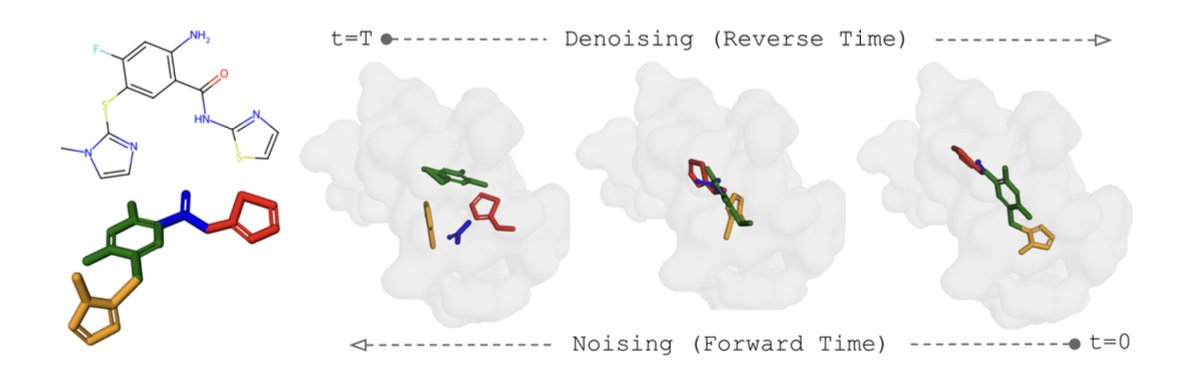
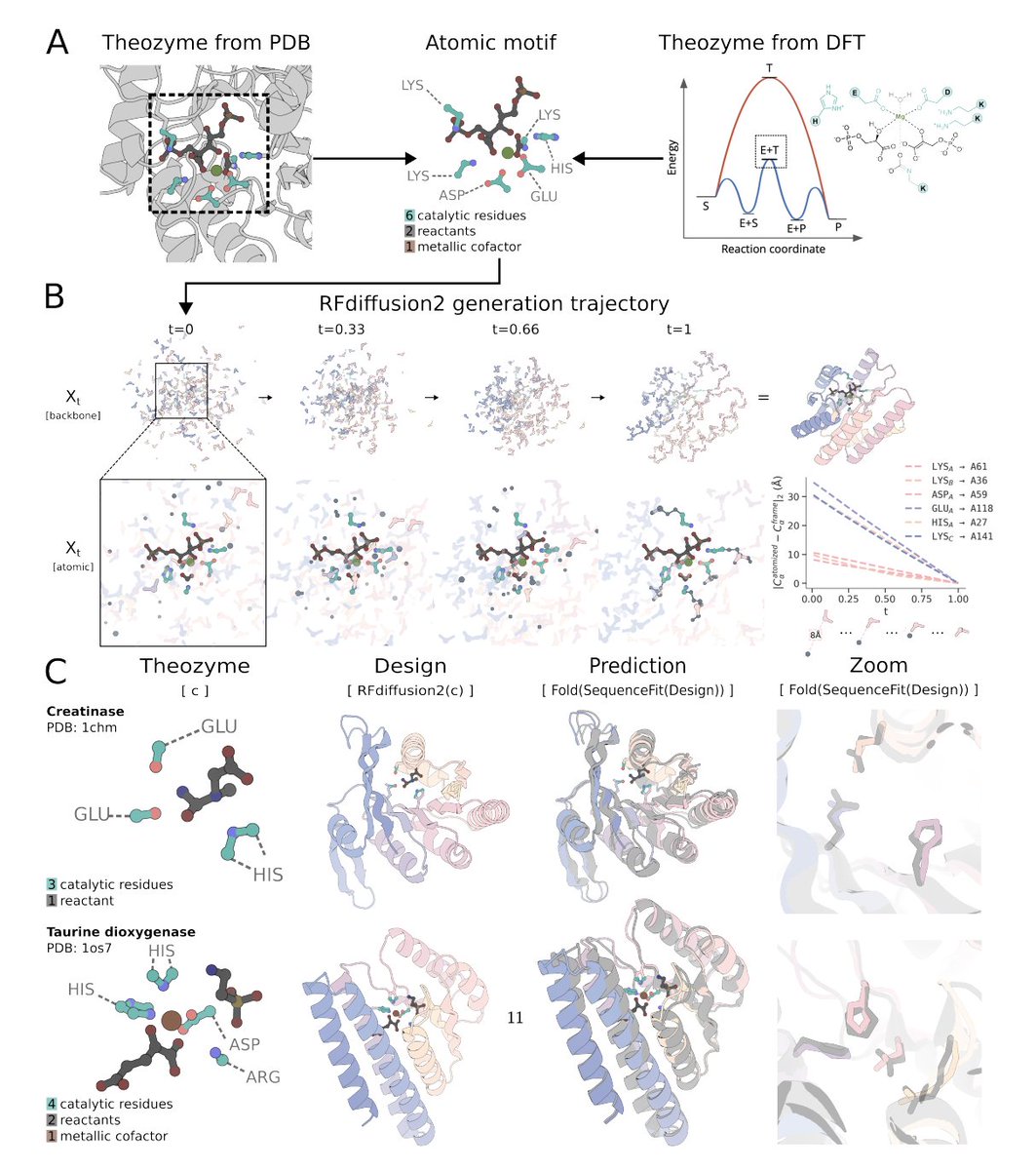
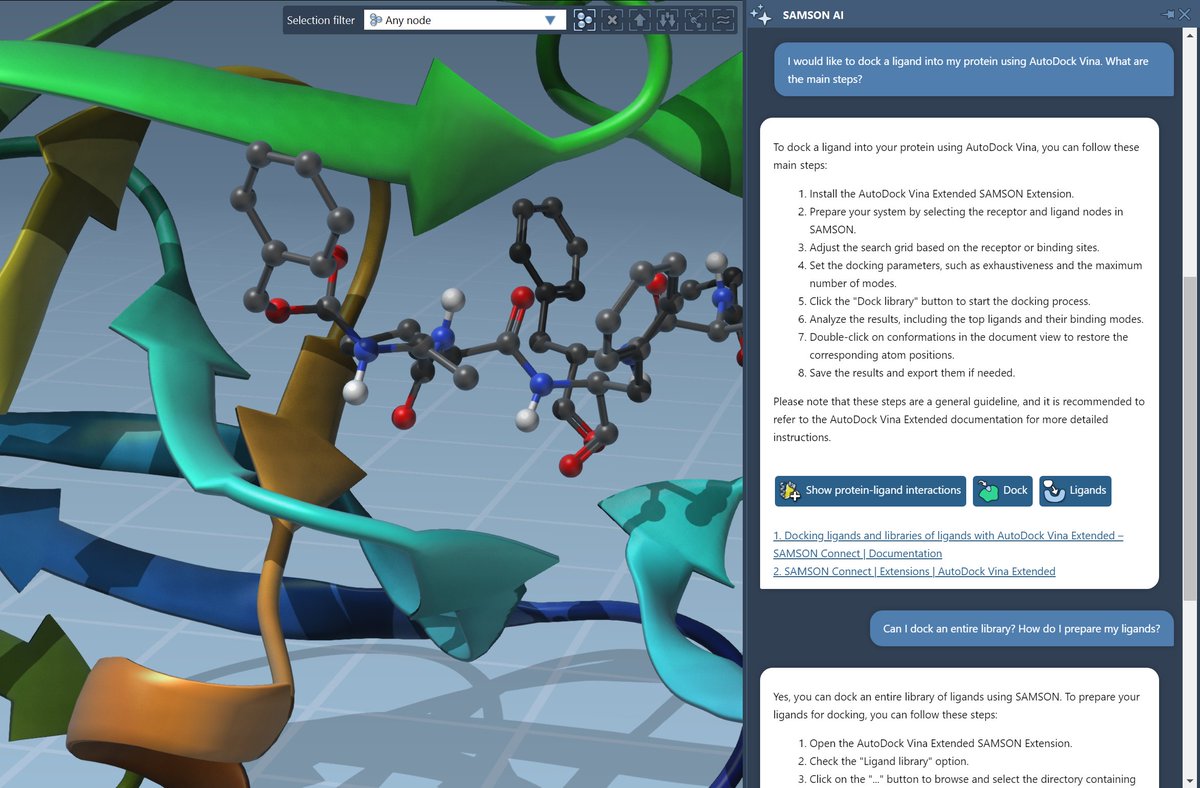
Our latest research, "An in vitro approach for simulating divergent Golgi O-glycosylation of tumour-associated MUC1 from normal MUC1," is published in Nature
Comms
nature.com/articles/s4146…
#CancerResearch #Glycobiology
#NatureCommunications #Innovation #Struenodo
English