sanj@splicewiring
We were curious whether the autoregressive next token prediction recipe that has worked for language can be applied to single-cell biology. We first tried to understand if a discrete representation is the right substrate for modelling in this domain? Tldr: Yes!
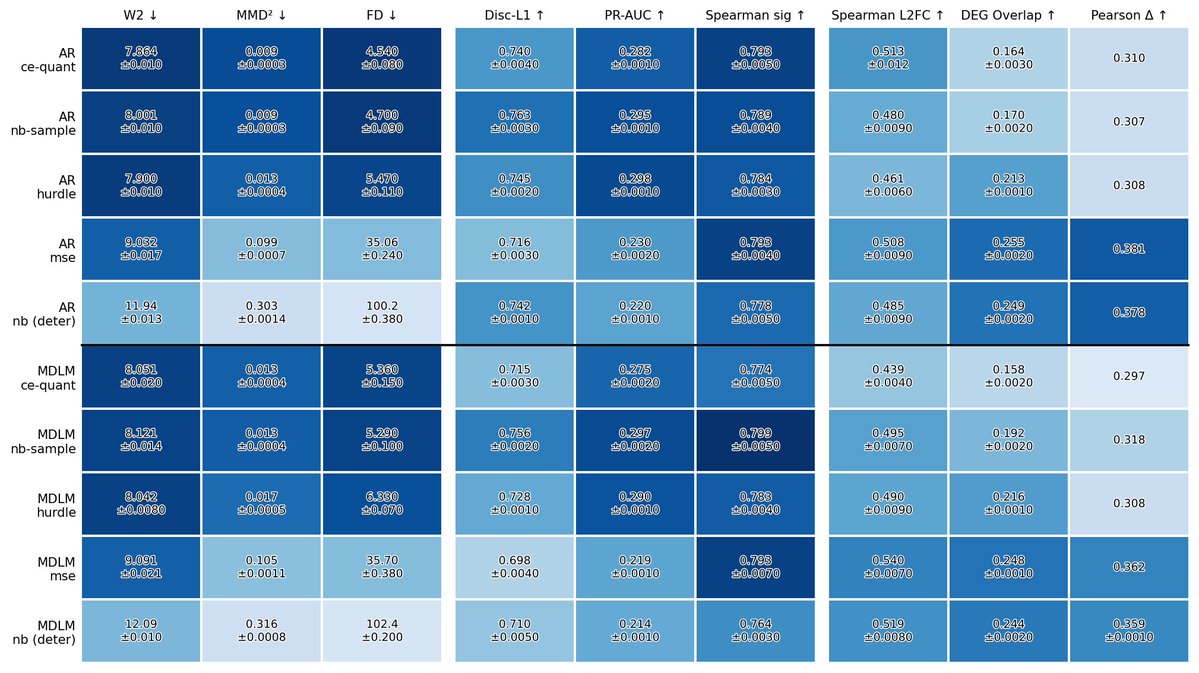
We studied a discrete latent recipe for training perturbation prediction models, that sets a new SOTA for most cell-eval metrics. We also asked: what does "best" even mean here?
There are mean-based metrics, which mostly ask whether you got the average perturbation effect right.
And there are distributional metrics, which ask whether you captured the shape and diversity of the predicted cell population.
As in, what is special about the models that perform on the mean based metrics class, as opposed to distribution based and vice-versa? (P.S. It’s not a mutual win in the literature we’ve found so far)
It turns out that a lot of this variance becomes clear when viewed through one axis: sampling richness. Some heads are better at collapsing toward a mean. Others are better at producing the distribution. Which one “wins” depends heavily on the ruler.
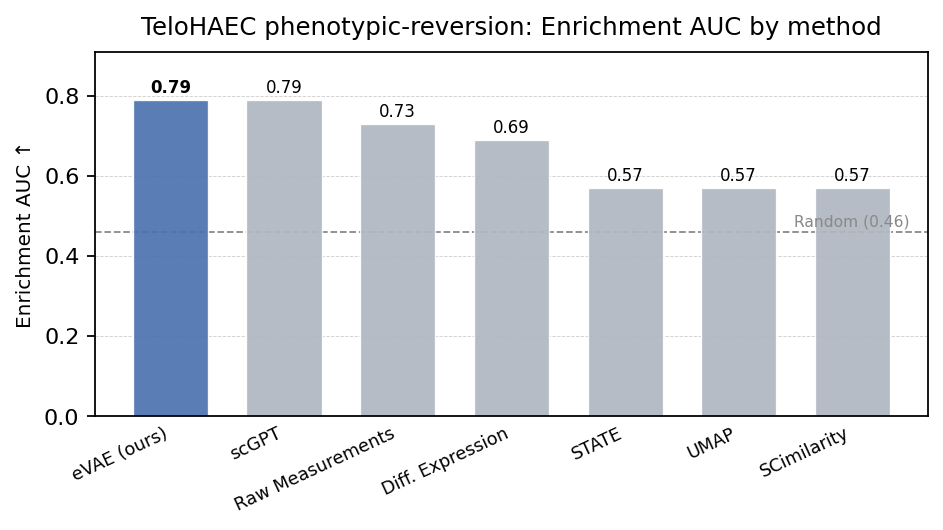
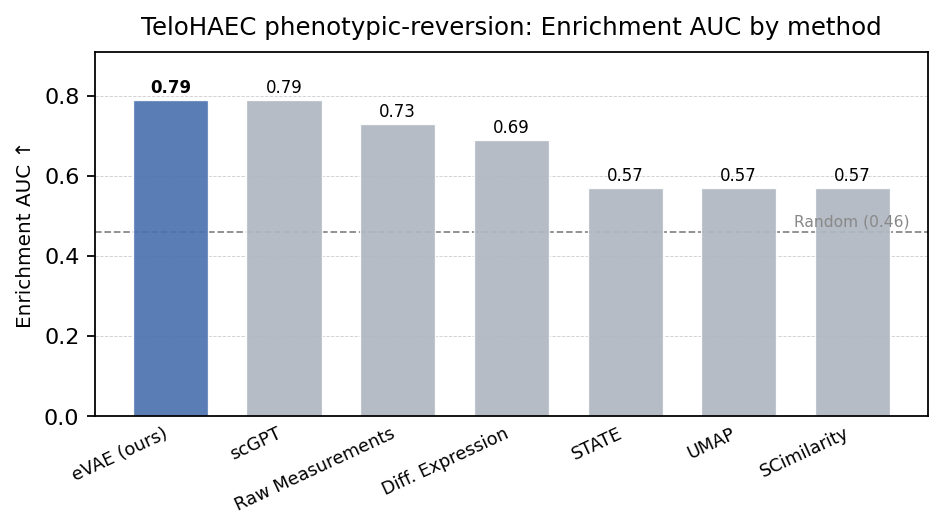
We then looked at a more biologically relevant task, where in a model is used to rank / predict something and then that’s reviewed from an expert and subsequently scored. On a held-out CRISPRi inflammation-reversion screen, the our encoder ranks target genes with 0.79 AUROC, matching scGPT while seeing roughly an order of magnitude less data. The benchmark carries several distribution shifts at once (cell type, perturbation set, biological context), and with a single dataset we cannot fully attribute the result to any one axis of generalization.
However, we hypothesize that this is because the models are trained on perturbational data and not just observational data.
cc: @Cgensbigler and @ShaamilKarim1