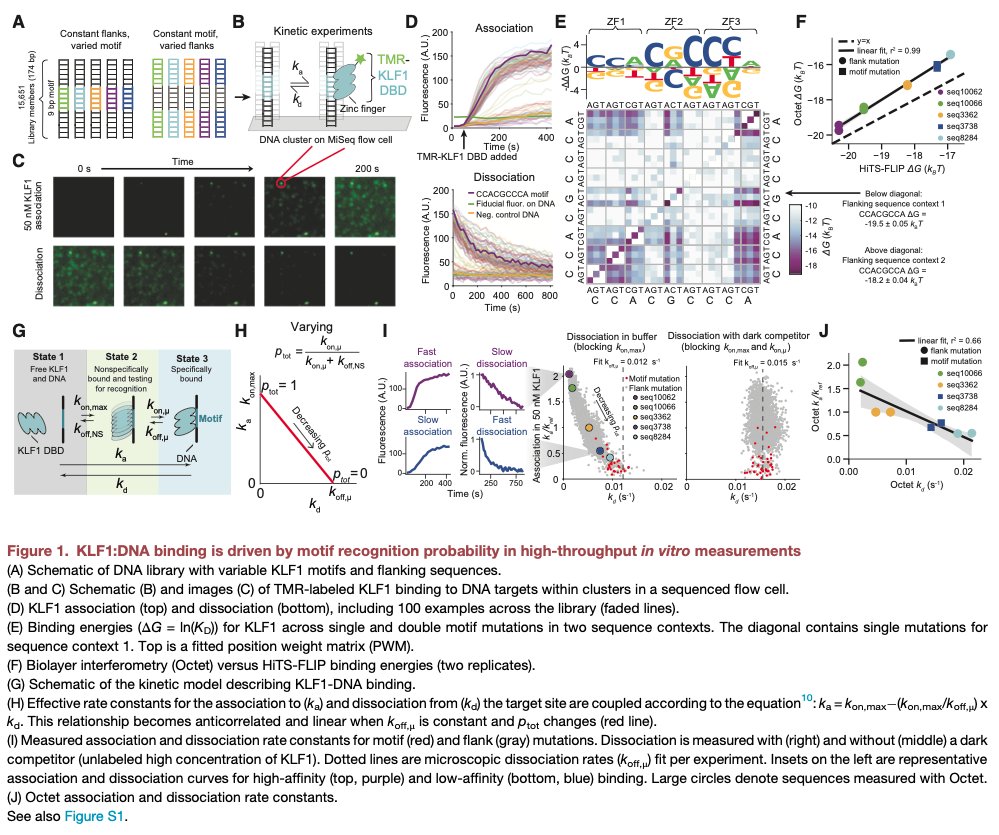
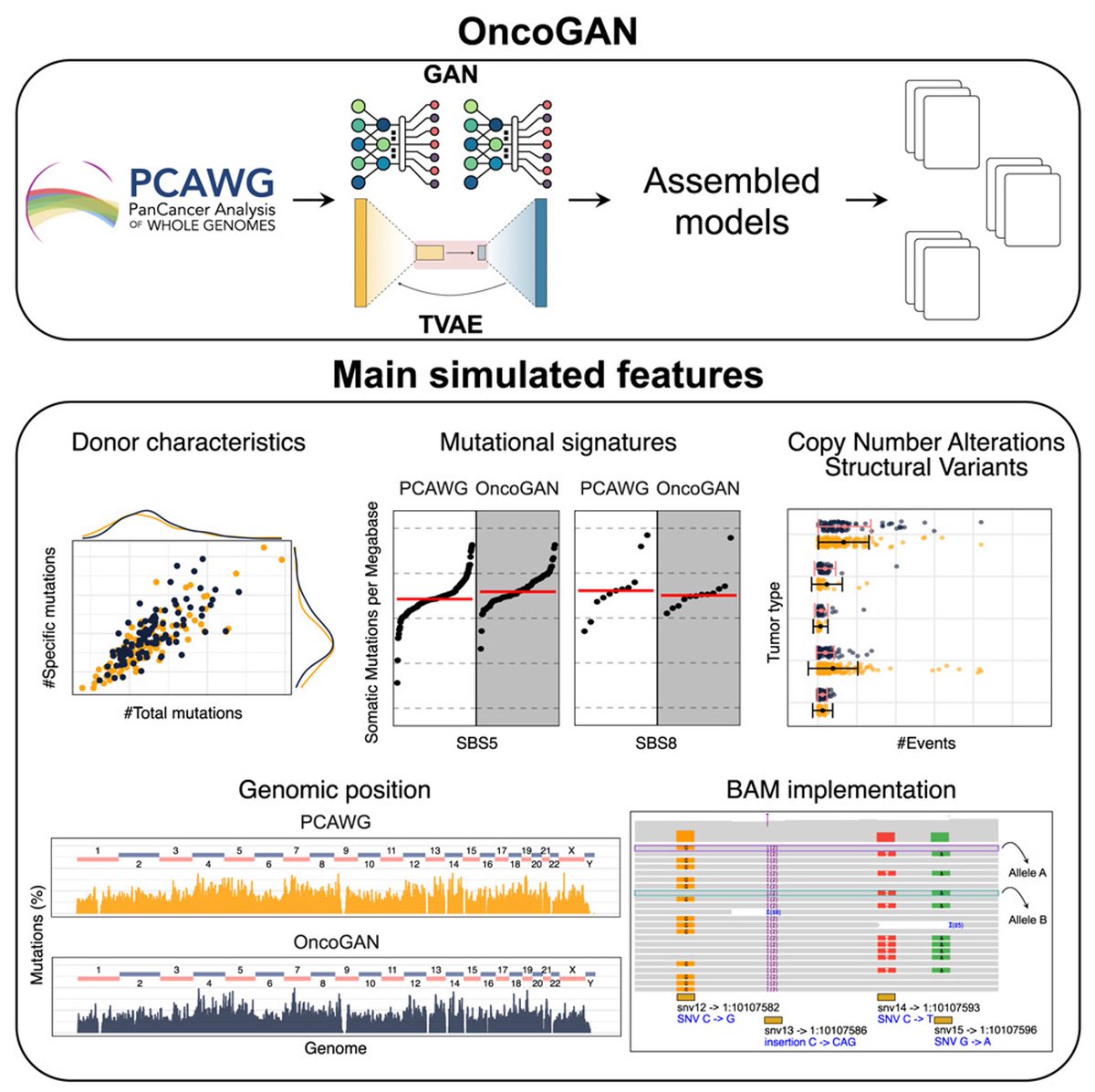
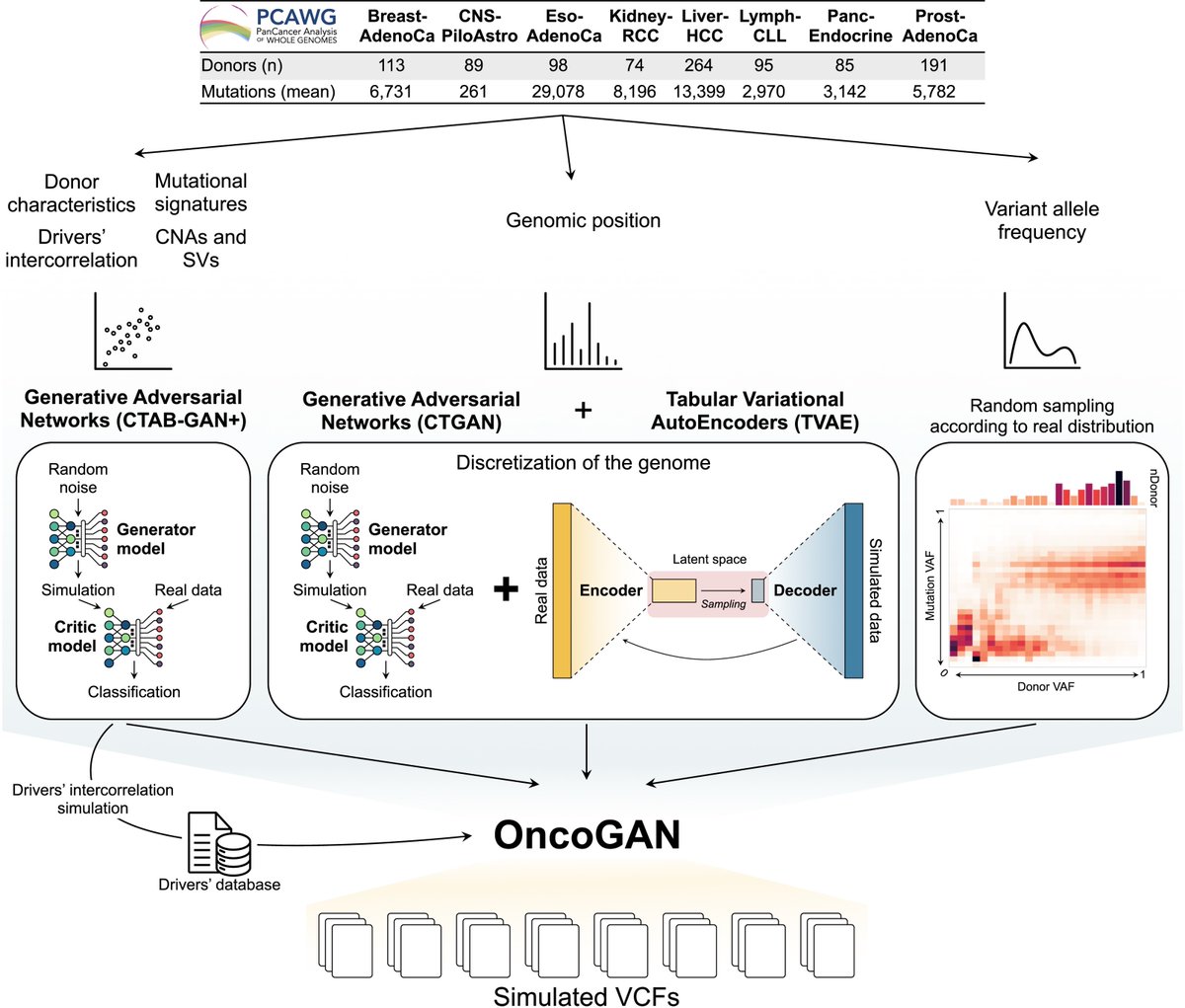
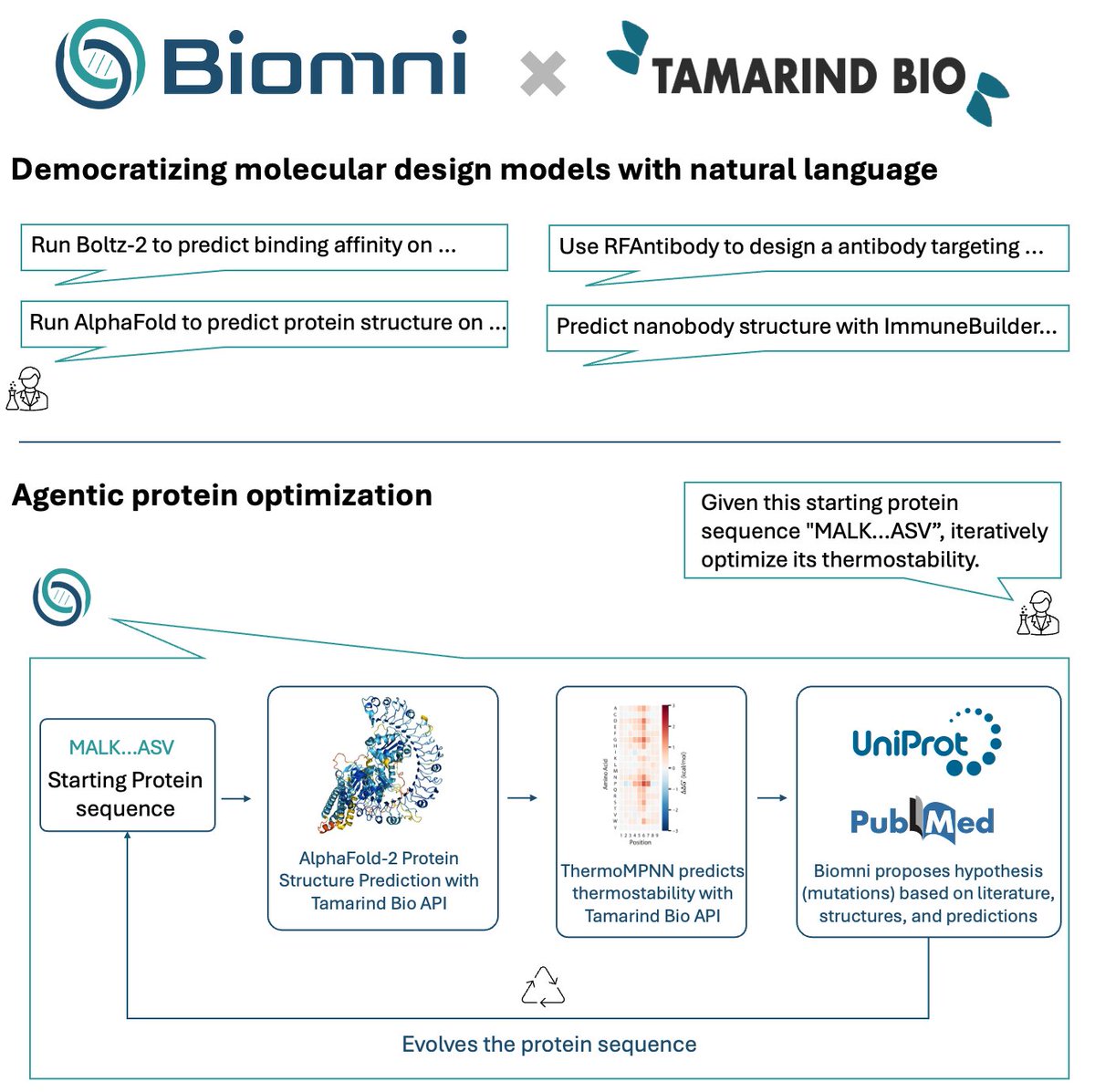
Pinned Tweet

Excited to share our work introducing EPInformer🧬, a scalable and lightweight deep learning framework to predict gene expression by integrating promoter-enhancer sequences with epigenomic signals and chromatin contacts. 📜biorxiv.org/content/10.110… (1/11)
English