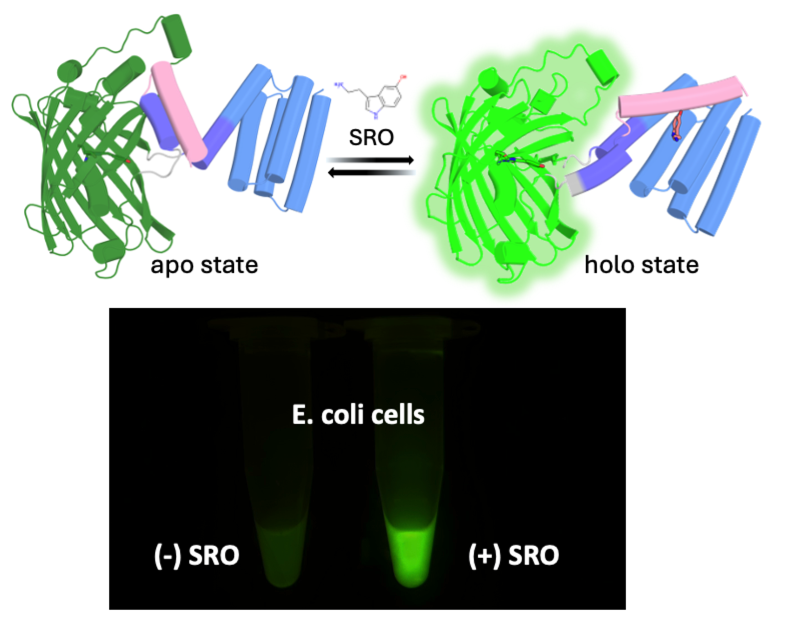
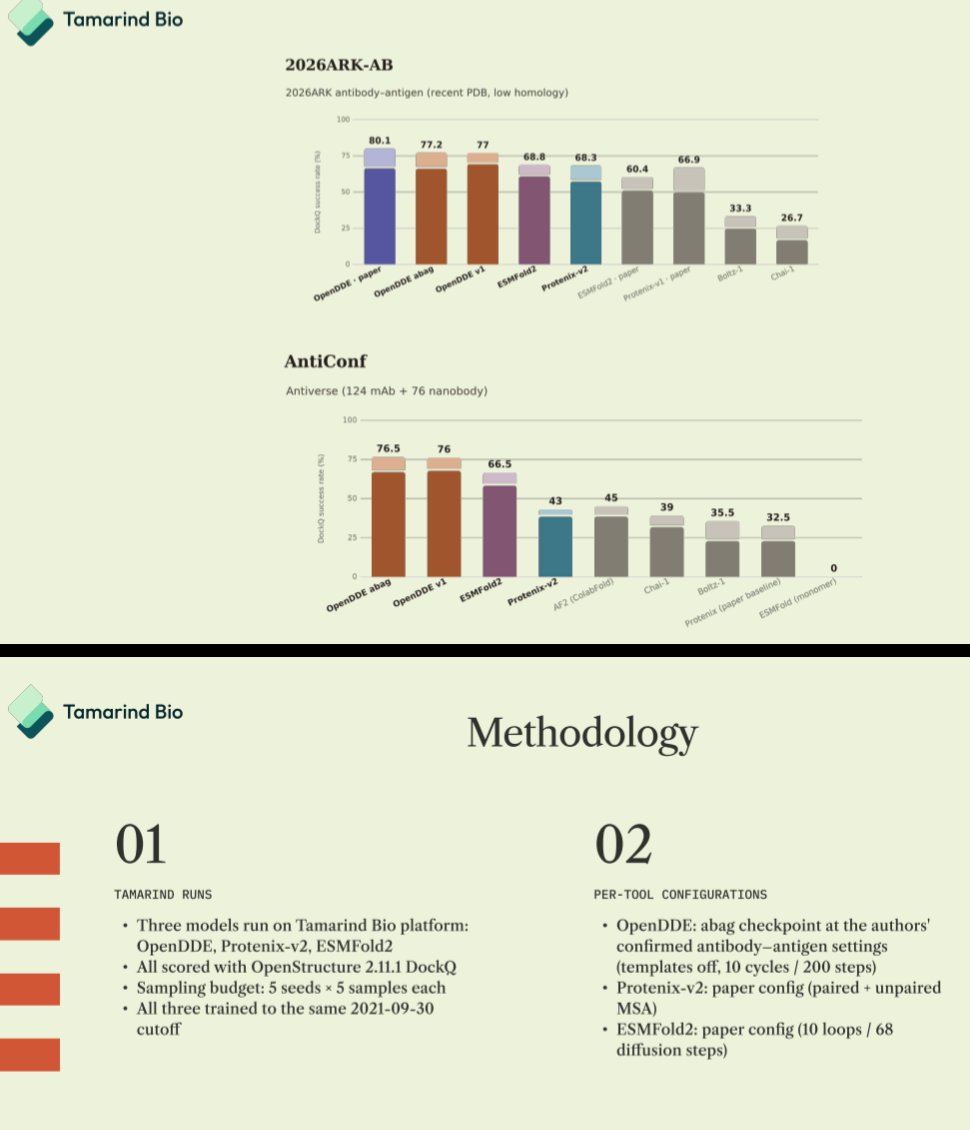
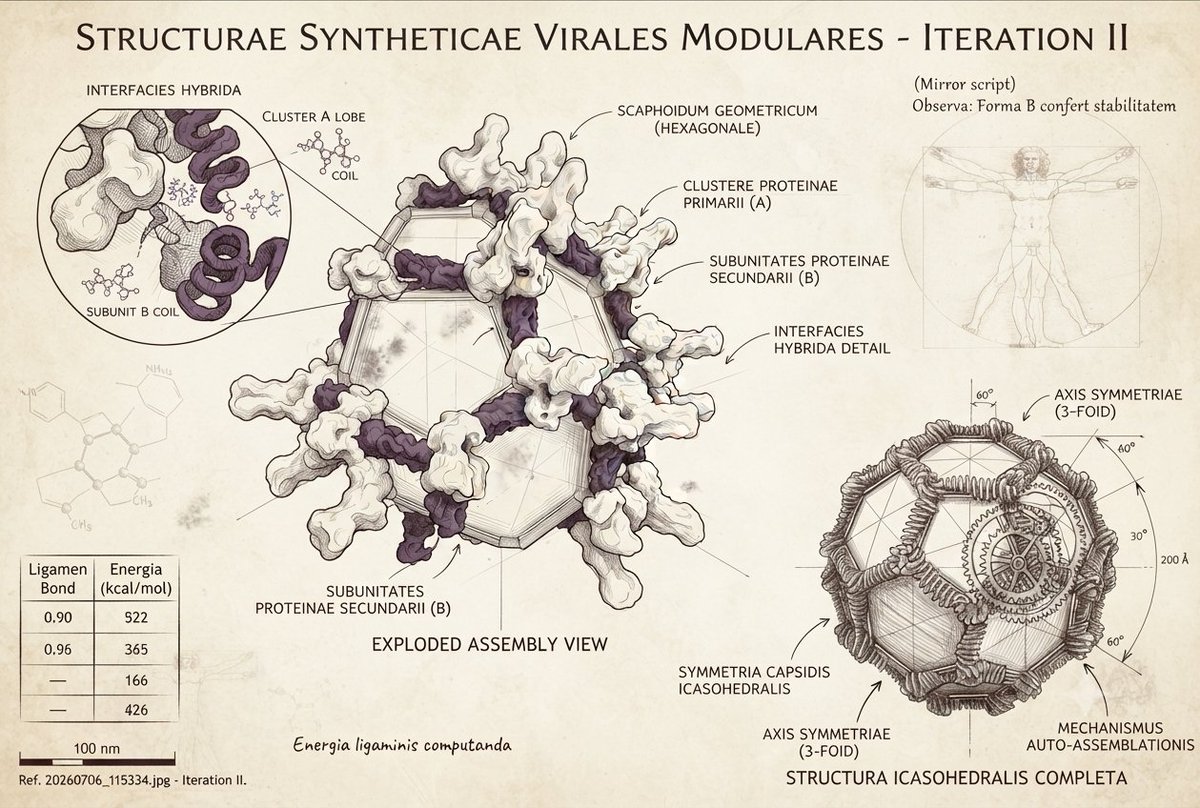
Biology+AI Daily@BiologyAIDaily
Vilya-1: An all-atom foundation model for macrocycle structure prediction and design
1. Vilya-1 is an all-atom diffusion-based foundation model aimed at a core bottleneck in macrocycle drug discovery: reliably sampling biologically relevant low-energy conformations across arbitrary, synthetically accessible chemistries (not just canonical peptides).
2. On 66 cyclic-peptide X-ray structures, Vilya-1 samples a near-native ring conformation (ring RMSD < 1 Å) in 89.2% of cases, vs 37.6% (Prime-MCS), 34.5% (RDKit ETKDGv3), ~15% (Boltz-2 / RF3), and 4.8–17.0% for several deep-learning conformer generators (TorDiff/LoQI/ETFlow).
3. The model generalizes across macrocycle classes/topologies, including disulfide-stapled peptides, sidechain-to-sidechain cyclizations, tail-to-sidechain cyclizations, and non-peptidic macrocycles (e.g., macrolides/polyketides). Reported examples include FK506, Sanglifehrin A, Lorlatinib, α-amanitin, and others with sub-Å ring accuracy.
4. A key design choice is a uniform heavy-atom representation that does not label “peptide vs small molecule,” enabling one architecture to cover mixed peptidic/non-peptidic scaffolds and even standard small molecules—addressing a common failure mode of residue-tokenized or protein-centric co-folding approaches on non-canonical chemistry.
5. Architecture-wise, Vilya-1 uses a single unified transformer for both representation learning and the diffusion process (rather than separate trunk + diffusion modules), with triangle attention/multiplication and pair-bias attention; inputs include atom scalar features, pairwise features, and atom vector features, and outputs 3D coordinates directly.
6. Training combines heterogeneous structural sources: public small-molecule and peptide crystal structures plus computationally generated macrocyclic peptide structures. Losses include diffusion coordinate reconstruction, a distogram auxiliary loss, and an explicit chirality loss to reduce stereochemical errors.
7. For receptor-bound conformations of macrocycles (240 PDB-derived ligands), Vilya-1 reaches 93% success (ring RMSD < 1 Å) despite not being trained on these structures; the paper notes that many entries in this benchmark appear in Boltz-2’s training set, highlighting Vilya-1’s emphasis on generalization rather than overlap.
8. The work adds a confidence model (fine-tuned from the conformer generator) to rank sampled conformers. On top-1 selection, confidence ranking improves success by 21.1% over random, comparable to MLIP energy-based scoring (21.9%), while being far cheaper computationally (~40 ms per conformer vs ~6120 ms for MLIP scoring with minimization).
9. Vilya-1 is also fine-tuned for multi-task property prediction from generated conformers (permeability via PAMPA/MDCK, chromatographic LogD, kinetic solubility, plus computed 3D polar surface area). Evaluation uses enrichment factor (top 10%) and includes time-based splits for internal data and scaffold splits for external datasets; structure-pretraining generally improves transfer, especially in “hit-series” settings where subtle conformational differences among close analogs drive permeability.
10. For design, Vilya-1 supports ligand-centric conformational landscape analysis: generating ensembles, scoring with MLIPs, and computing Pnear to quantify preorganization. Across three campaigns, prioritizing by Pnear enriches binders (Kd ≤ 50 µM) without modeling the target explicitly—suggesting conformational preorganization alone can be a useful, target-agnostic filter.
11. The paper also demonstrates discrete multi-objective optimization to “miniaturize” larger macrocycles by scaffolding key binding motifs into smaller rings (e.g., shrinking 13–14mers to 7mers) while optimizing predicted developability (higher permeability/hydrophobicity, lower solvent-accessible polar surface area), and it explicitly supports complex cyclization chemistries used in display technologies (thioether, γ-lactam, i,i+7 staples).
📜Paper: arxiv.org/abs/2607.09998
#ComputationalChemistry #Macrocycles #CyclicPeptides #DiffusionModels #MolecularModeling #DrugDiscovery #Cheminformatics #StructuralBiology #MachineLearning