Sabitlenmiş Tweet
イージーコー
9.7K posts

イージーコー
@e_zyko
日本に住んでいます〜 travel | coffee | gourmet | Editorial board of Sci Rep
日本 Katılım Aralık 2020
294 Takip Edilen733 Takipçiler

イージーコー retweetledi

อยู่ญี่ปุ่นมาซักพักละ ตกผลึกได้แบบนี้
สำหรับคนมาเรียน
ถ้าได้ทุนมาเรียน อันนี้ตามสบายเลย แต่ถ้ามีความสามารถก็ลองหาทุนประเทศอื่นด้วย คิดว่าไม่ยากเกินไป
ถ้าออกทุนเอง อันนี้ขอสงวนความเห็น
สำหรับคนมาทำงาน ถ้าจะย้ายมาญี่ปุ่นเพราะอยากมาเก็บประสบการณ์หรือว่าอยากมาลองใช้ชีวิตต่างบ้านต่างเมืองก็คิดว่าเป็นตัวเลือกที่ดี โดยเฉพาะถ้ามาทำงานในบริษัทข้ามชาติที่ไม่ญี่ปุ่นมาก หรือว่างานสาย tech ก็จะปรับตัวได้ง่ายหน่อย ถ้าไม่ใช่ก็ต้องสตรองระดับนึง
แต่ถ้าจะมาเพราะเรื่องเงินก็ไปที่อื่นเหอะ
sgtubaby@sgtubaby
ใครที่ย้ายประเทศหรือไม่ได้กลับไทยเกิน 1 ปีแล้ว มีคำแนะนำสำหรับคนที่อยากย้ายประเทศไหมครับ
ไทย

ใครเคยไป KL บ้าง ถ้าไม่พกเงินสดไปเลยอยู่ได้ไหม แค่ไปทำงาน 2 วัน ไม่ซื้ออะไรเลย เน้นกินเพื่ออยู่ ถ้ามีแค่ true money กับ บัตร mc 1 ใบ อยู่รอดไหม ในการซื้อตั๋วรถไฟ เข้าเมือง ไปห้าง ซื้อของ ร้านสะดวกซื้อ
ไทย
イージーコー retweetledi


@wwaterfall96 คำว่า"ไม่รัก"จังหวะมันเสียงดังหรือน้องๆออกเสียงไม่ค่อยชัด(แต่เพลงฟังดูน่าสนุกดีนะคะ)☺️
ไทย

อหหหหหหหหหห เพลงในโปรเจค the test of time ของแอทลาสคือเพลงโทรมาทำไม กิมมิคคือท่อนแรปมีเปลี่ยนเป็นชื่อเมมเบอร์ girl group t-pop มีทั้ง 4EVE PiXXiE ALALA SNS DIDIxDADA Ally-Jetaime เจนิส เจนเย่ เป็นต้น กุให้เลยอะ ชอบมาก โดนเส้นค่ดดดดด 😭😭😭😭😭😭
#SunsationalSongkranFestivalxATLAS
#ATLAS_TH
ไทย
イージーコー retweetledi

Fast sampling of protein conformational dynamics @ScienceAdvances
1. Sauer et al. show that the key collective variables (CVs) needed to drive enhanced sampling of protein conformational transitions are encoded in anharmonic low-frequency vibrations, and these CVs can be extracted from short unbiased MD without any prior knowledge of the transition.
2. Core idea: use FRESEAN (frequency-selective anharmonic mode analysis) at (near) zero frequency to isolate collective motions with minimal restoring forces—i.e., “paths of least resistance” for conformational change—avoiding the limitations of harmonic/quasiharmonic normal modes in the low-frequency, diffusive regime.
3. Practical pipeline: run 20 ns unbiased all-atom MD, align trajectories, coarse-grain to a 2-bead-per-residue representation (1 for Gly), compute velocity time-correlation matrices, Fourier transform to frequency domain, then take eigenvectors at zero frequency. Modes 1–6 correspond to translation/rotation and are discarded; modes 7+ capture internal anharmonic low-frequency vibrations.
4. Reproducibility is a central result: across 5 independent 20 ns replicas per protein, the low-frequency modes (especially the 2D subspace spanned by modes 7–8) are consistently recovered, unlike PCA/quasiharmonic modes whose replica-to-replica agreement remains poor even with much longer trajectories.
5. Enhanced sampling step: use modes 7 and 8 as CVs in well-tempered metadynamics (100 ns per run; reported as <24 hours on a single GPU). Across 5 proteins × 5 replicas, 22/25 runs (88%) sample known “closed↔open” transitions within 100 ns; extending to 160 ns yields full sampling for all replicas.
6. Benchmark set spans diverse challenges: HEWL (disulfide-stabilized), HIV-1 protease (homodimer), MCL-1 (allosteric/druggable dynamics), ribose-binding protein (multi-domain hinge motion), and GDP-bound KRAS (switch-region dynamics). The same FRESEAN-to-metadynamics protocol is applied across all systems.
7. Free-energy landscapes (FES) become both fast and statistically controlled by running 20 parallel metadynamics replicas (20 × 100 ns) using the same FRESEAN CVs: single-run uncertainties are typically < ±10 kJ/mol, and averaging reduces standard error to < ±3 kJ/mol, enabling reproducible thermodynamic ensembles rather than just qualitative transitions.
8. Comparison to “hand-crafted” geometric CVs from prior literature is informative: biasing along FRESEAN modes often follows lower-free-energy transition routes and tends to keep sampling within the native folded ensemble, whereas geometric CVs can push systems into partially unfolded high-entropy states (most notably KRAS when biased by residue–residue distances).
9. The authors quantify cross-CV reweighting fidelity using Shannon entropy and Bhattacharyya coefficients: on average, ensembles generated by biasing along low-frequency vibrational CVs preserve at least as much (often more) information when reweighted into geometric-variable space than the reverse, supporting the claim that these vibrations are broadly suitable, system-agnostic CVs.
10. Implication for computational biology/ML: the method enables high-throughput generation of conformational ensembles and FESs (including mutants/conditions), helping address the dataset bottleneck for next-generation sequence→structure→dynamics models beyond single static folds or single thermodynamic states.
💻Code: github.com/HeydenLabASU-c…
📜Paper: doi.org/10.1126/sciadv…
#MolecularDynamics #EnhancedSampling #Metadynamics #ProteinDynamics #FreeEnergy #ComputationalBiophysics #CollectiveVariables #FRESEAN #GROMACS #PLUMED

English
イージーコー retweetledi

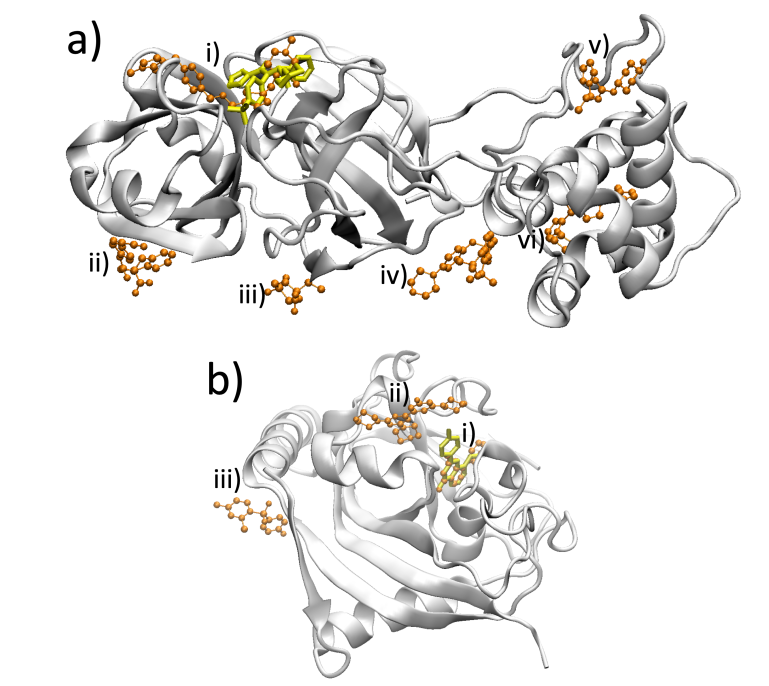
Boltz-2 for drug discovery - is the hype justified?
A new study tested it on 38,000+ compounds across two targets:weak to moderate correlation with physics-based binding free energies. Among the top 100 compounds: no significant correlation at all.
arxiv.org/abs/2603.05532
English
New in DFDD: Direct Absolute Binding Free Energy (DBFE) support is now included.
This update broadens the DFDD workflow by adding binding free energy analysis.
Link to DFDD: tinyurl.com/4er32apn
Learn more about DBFE here: github.com/molecularmodel…

English
Run MD simulation on your iPad or Cloud, no install needed! 🚀
Fully browser-based
Auto-analysis: RMSD, Free Energy, & publication-ready plots~
For education and research!
#compchem #MDsimulation #chemcom #bioinfomatics
@JCIM_JCTC asap!
pubs.acs.org/doi/10.1021/ac…
English
イージーコー retweetledi

#HighlightOfTheWeek Trajectories showing ligand insertion into the beta-cyclodextrin cavity.
#compchem
pubs.acs.org/doi/10.1021/ac…
English
イージーコー retweetledi

🚨 BREAKING: Claude can now research like an MIT PhD student.
Here are 12 insane Claude prompts that turn 40+ research papers into structured literature reviews, knowledge maps, and research gaps in minutes.
(Save this before it goes viral):

English
イージーコー retweetledi

🚨 BREAKING: You can now run full drug discovery pipelines inside Claude Code with one command.
Claude-Scientific-Skills is an open-source plugin that gives Claude Code 140 ready-to-use scientific workflows with a single install. It covers biology, chemistry, medicine, and ML without manual configuration.
→ Connects to 28+ databases like PubMed and ChEMBL.
→ Run drug discovery pipelines and molecule docking.
→ Analyze raw single-cell data and map gene networks.
→ Cross-reference clinical variants and drug interactions.
100% Open Source.
English
イージーコー retweetledi

My powerpoint slide on the plant virus I am working on 🌱🧬
English
Transparency and acknowledgment are essential for cumulative science.
Respect frameworks, credit origins, and cite properly.
#AcademicIntegrity #EthicalPublishing #OpenScience
English
COPE and Nature remind us:
“Ideas, methods, and presentation formats require proper attribution—
not just copied text.”
> publicationethics.org/guidance/Guide…
> nature.com/articles/s4159…
English
Plagiarism isn’t just about copying text.
A growing issue in research is copying methodological frameworks or graphic styles without citation — a silent breach of academic ethics.
#ResearchIntegrity #AcademicEthics
English
イージーコー retweetledi

นางเอกที่น่าสงสารที่สุดในละครไทย เด็กมปลายแต่งงานใช้หนี้เจ้าสัวที่มีเมีย 3 คนในบ้าน ดันมารักลูกชายเค้า ตอนจบเห็นเจ้าสัวจับคุณนายที่ 3 ถ่วงบ่อหลังบ้าน ตกใจวิ่งหนีหาพระเอก เห็นพระเอกกับเพื่อนเกย์นอนด้วยกัน สติขาดผึง เป็นบ้า เหมือนเอาทั้งชีวิตวัยสาวมาทิ้ง สนุกกว่าดอกส้มสีทองเพราะเล่าจากมุมมองเมีย 4 คนในบ้าน
มุยยิมฟงเกลียดคนแดกภาษี@loveteacher_law
สิ่งที่สยองที่สุดคือคุณนายที่4 อายุประมาณ15-16 นี่แหละถ้าจำไม่ผิด แล้วมันจะมีฉากนึงที่กินข้าวรวมกันทั้งครอบครัวคุณนายที่4 ใส่ชุดนักเรียนคอนแวนต์ด้วยน่าจะกินข้าวก่อนไปโรงเรียนมั้งเพราะพวกลูกๆเจ้าสัวก็ใส่ชุดนักเรียนเหมือนกัน จะอ้วกอีเจ้าสัวจ้าา🤢
ไทย
イージーコー retweetledi

イージーコー retweetledi
Drug discovery is slow, expensive, and uncertain. On average, it takes 12–15 years and billions of dollars to bring a new therapy to patients. Out of thousands of molecules, maybe one makes it all the way.
This is where computational methods like Molecular Docking are changing the game.
In our latest blog, @TensorTwerker breaks down the fundamentals of docking in drug discovery, not just what it is, but how it actually works under the hood.
🔹 What you’ll learn:
> How docking predicts both binding poses and binding affinity
> The algorithms that power docking (genetic algorithms, Monte Carlo, systematic searches)
> The role of scoring functions (physics-based, empirical, knowledge-based)
> Different types of docking (rigid, flexible, protein–ligand, protein–protein, protein–nucleic acid)
> Docking workflows: from protein/ligand prep to evaluation
> A look at commonly used software (AutoDock, Glide, GOLD, DOCK6, HDOCK, etc.)
> The limitations: protein flexibility, solvation, scoring function accuracy, and the GIGO problem
> How docking fits into a broader pipeline with molecular dynamics, FEP, and experimental validation
Do give it a read. Links in the comment
Anindyadeep@anindyadeeps
In our recent blog, @TensorTwerker dives deep to help you understand about molecular docking in drug discovery. Links in the comments. We discuss topics about how docking works under the hood, different search and scoring algorithms, types of docking (protein–ligand, protein–protein, protein–Nucleic Acid), common software, and the real limitations you need to be aware of. If you’re working in drug design or just curious about how computational biology is reshaping pharma R&D, or interested in AI x Science, this one’s worth a read.
English