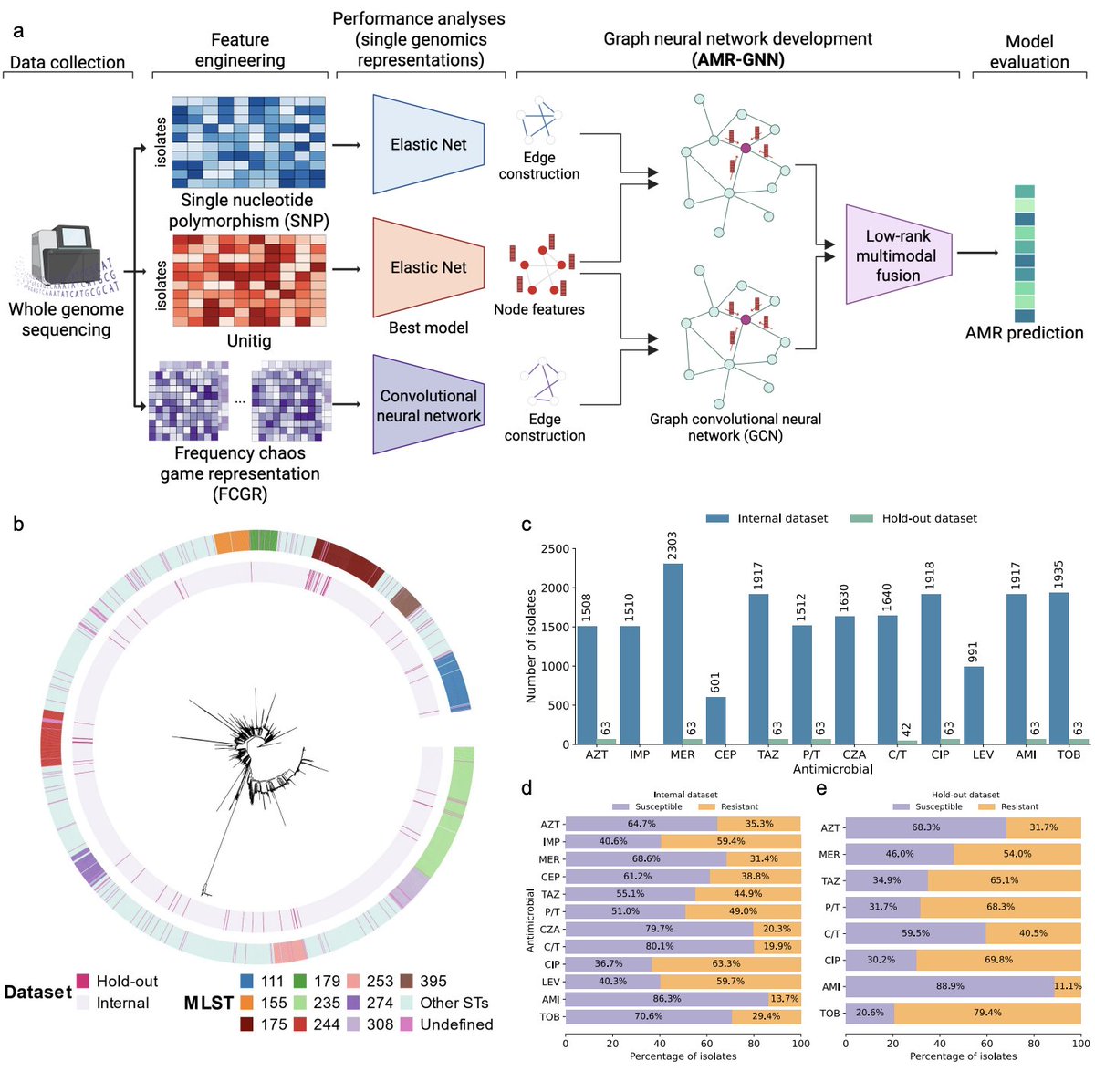
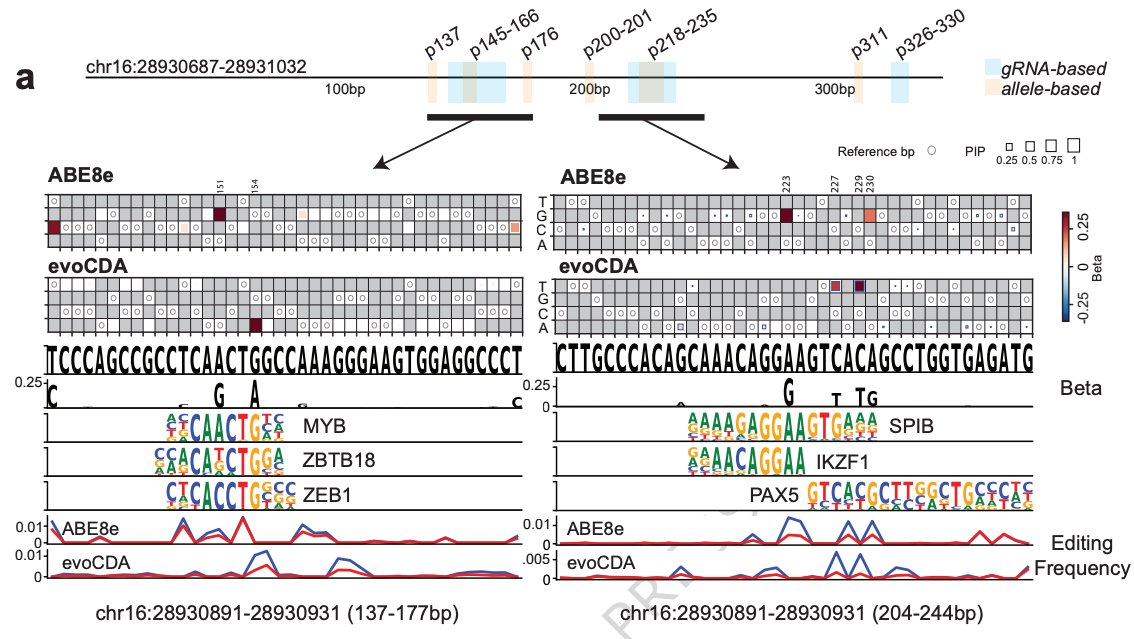
Reposting: My lab has an open postdoctoral position to lead large-scale gene association and PheWAS in major biobanks and develop AI/ML models to predict genetic variant function. Strong programming skills, a solid publication record, and human genetics background are required.
English